


- Accueil
- Volume 14 (2010)
- numéro 4
- Comparison of the glucooligosaccharide profiles produced from maltose by two different transglucosidases from Aspergillus niger


Visualisation(s): 3966 (18 ULiège)
Téléchargement(s): 265 (1 ULiège)

Comparison of the glucooligosaccharide profiles produced from maltose by two different transglucosidases from Aspergillus niger

Notes de la rédaction
Received on July 16, 2009; accepted on December 1, 2009
Résumé
Comparaison des profils en glucooligosaccharides produits à partir de maltose par deux transglucosidases différentes d’Aspergillus niger. Les préparations prébiotiques d’isomaltooligosaccharides (IMO) contiennent des α-D-glucooligosaccharides et la structure de ceux-ci est l’élément majeur déterminant le potentiel prébiotique d’un produit. Il est établi que la sélectivité de la réaction de transglucosylation dépend de la spécificité de l’enzyme et que le maltose et les α-glucooligosaccharides peuvent à la fois jouer le rôle de donneur ou d’accepteur de résidus glucosyl au sein de la réaction. Deux enzymes commerciales, une glucosyltransférase et une α-glucosidase, ont donc été testées seules ou en combinaison sur du maltose pur afin d’étudier leurs spécificités et le profil en IMO obtenu. Les réactions ont été suivies à l’aide d’une méthode analytique améliorée AEC-PAD permettant la détection et la résolution de nouveaux IMO inconnus. La détermination structurale de ces nouvelles espèces a ensuite été entreprise par le biais de leur temps de rétention et leur abondance relative. De manière générale, l’α-glucosidase possède une activité hydrolytique plus prononcée produisant des composés contenant moins de liaisons résiduelles α-(1-4) digestibles telles que l’isomaltose, l’isomaltotriose, l’isomaltotétraose, le kojibiose et le nigérose, alors que la glucosyltransférase produit des quantités importantes de panose. Enfin, la combinaison des deux enzymes a permis l’obtention d’un profil en IMO intermédiaire. La composition des sirops d’IMO s’avère donc dépendante de la spécificité de l’enzyme transglucosylante, ce qui permet de faire varier le profil en IMO du produit obtenu en jouant sur le choix des enzymes et leur proportion.
Abstract
Prebiotic isomaltooligosaccharide (IMO) preparations contain α-D-glucooligosaccharides and their structure is the key factor for their prebiotic potential. The transglucosylation selectivity is known to depend on the enzyme specificity and moreover, maltose and α-glucooligosaccharides can actually act as both glucosyl donor and acceptor in the reaction. Thus, two commercial enzymes, a glucosyltransferase and an α-glucosidase, were tested alone and in combination on pure maltose to study their specificities and the IMO profile obtained. The reactions were monitored using a step-forward AEC-PAD analytical method which permitted to detect and resolve new unknown IMO. Structural determination of unknown IMO was attempted using their retention times and relative abundance. As a general rule, the α-glucosidase has a more expressed hydrolyzing activity leading to products containing less residual digestible α-(1-4) linkages such as isomaltose, isomaltotriose, isomaltotetraose, kojibiose and nigerose while the glucosyltransferase produces important amount of panose. Finally, the combination of the two enzymes leaded to an intermediate IMO profile. IMO syrups composition was thus proved to be dependant on the specificity of the transglucosylating enzyme so that products profiles can be designed using different enzymes and in different proportion.
Table des matières
1. Introduction
1Prebiotics are non-digestible dietary components that pass through the digestive tract to the colon and selectively stimulate proliferation and/or activity of desired populations of bacteria indigenous to the human or animal colon in situ (Gibson et al., 1995; Rycroft et al., 2001; Roberfroid, 2008; Wang, 2009). Among these prebiotics, isomaltooligosaccharides (IMO) are for years the most successful ones in Asian countries thanks to their many favorable properties for application in food industry (Kohmoto et al., 1988; 1991; Kanno, 1990; Kaneko et al., 1995; Gu et al., 2003; Goffin et al., 2010).
2Prebiotic mixtures composition is the key factor for their efficiency throughout the structure/function relationship as well as the possible partial digestion by endogenous enzymes that can occur before reaching the colon (Sanz et al., 2005). Consequently, the starting substrate, enzyme type and specificity together with the enzymatic reaction precise control are critical points to produce optimized IMO preparation with high prebiotic power. Two types of transglucosylating enzymes are available to produce IMO starting from hydrolyzed starch: glucosyltransferases (EC. 2.4.1.24) and α-glucosidases (EC. 3.2.1.20) (Plou et al., 2007). Glucosyltransferases catalyze group-transfer reactions of monosaccharides even in dilute reaction conditions. Transferase producing IMO are non-Leloir enzymes that require neither co-factors nor activated substrates, as they directly employ the free energy of oligosaccharides osidic bond cleavage (Plou et al., 2002). Glycosidases are hydrolytic enzymes, which can catalyze at a certain extent either reverse hydrolysis (thermodynamic control) or transglucosylation (kinetically-controlled process) (Perugino et al., 2004), since they are retaining glucosyl hydrolase (GH) (Chiba, 1997). Some α-glucosidases indeed exhibit clear transglucosylation activity (Kato et al., 2002; Faijes et al., 2007). In both cases, the non-reducing D-glucosyl residue of the donor may be transferred to water (hydrolysis), to D-glucosyl residues released by hydrolysis, or to the non-reducing residue of maltose and every α-glucooligosaccharide present in the solution. This transfer can occur to the 6-OH, 2-OH, 3-OH and/or 4-OH group of the non reducing glucose unit of the acceptor depending on the enzyme specificity. Maltose and α-glucooligosaccharides can actually act as both glucosyl donor and acceptor in the reaction. The starting substrate is, thus, also determinant because of the enzymes differential capacity to use various substrates as donor or acceptor in the reaction (Pazur et al., 1978; Hamada et al., 1980; Benson et al., 1982; McCleary et al., 1989). IMO preparations are thus composed of a complex mixture of glucose oligosaccharides characterized at the same time by their DP value (from 2 to ~ 10), linkages types [α-(1-2, 3, 4 or 6)] and the proportion and position of each type of linkage. Numerous articles since the 1950s, evaluated the production of IMO (Mc Cleary et al., 1989; Duan et al., 1994; Yun et al., 1994) and associated α-glucooligosaccharides (Yamamoto et al., 2004) by enzymes with specific activities using mainly paper chromatography (Barker et al., 1953; Nishi et al., 1975; Kato et al., 2002) and paper electrophoresis (Takahashi et al., 1988), or HPLC on amino column with RI (Duan et al., 1995; Nakanishi et al., 2006), ELSD (Fernandez-Arrojo et al., 2007) or UV with ABEE-labeled oligosaccharides (Kobayashi et al., 2003) detection. However, the first two methods do not permit the full discrimination of all structural isomers and their quantification while amino column resolution is still very limited. Consequently, results do not reflect the full complexity of the mixtures and quantification can be biased. It is important to specify that in this paper, IMO definition in the strict sense [only α-(1-6) linkages] is extended and IMO are considered as α-glucooligosaccharides possessing at least one “ALO” linkage (Anomalously Linked Oligosaccharides, Takaku, 1988) i.e. α-(1-6), α-(1-2) or α-(1-3). Indeed, these compounds are believed to participate to the prebiotic effect and are often considered in nutritional studies (Kaneko et al., 1994).
3Therefore, in this paper, two commercial enzymes, a glucosyltransferase and an α-glucosidase, are tested alone and in combination on pure maltose to study their activities and the IMO profile obtained. In order to monitor more accurately IMO production, a step-forward analytical method using AEC-PAD and able to discriminate structurally close molecule is used (Goffin et al., 2009a). Indeed, this method enables to separate a wider range of IMO and non-IMO by-products than previous methods (Koizumi et al., 1989; Ammeraal et al., 1991; Paskach et al., 1991; Vinogradov et al., 1998; Dols-Lafargue et al., 2001; Demuth et al., 2002; Nakanishi et al., 2006) and thus to detect new types of IMO and obtain a better picture of the product prebiotic potential. Moreover, some structures are proposed for these unknown IMO according to their retention times and relative abundances. However these structures will need to be confirmed individually in a coming paper by structural determination methods such as NMR or LC-MS.
2. Experimental
2.1. Reagents
4To prepare the standard solutions, sample solutions and the mobile phase, 18 MΩ purified water produced with a laboratory water purification system (Barnstead, IA, USA) was used throughout the experiments. D-Glucose, maltose and panose were purchased from Sigma-Aldrich SA (Bornem, Belgium), isomaltotriose, isomaltotetraose, isomaltopentaose, isomaltohexaose and isomaltoheptaose from Medac GmbH (Wedel, Germany), kojibiose, nigerose, maltotriose, maltotetraose, maltopentaose, maltohexaose and maltoheptaose from Wako chemicals GmbH (Neuss, Germany), nigerotriose from Dextra Laboratories LTD (Reading, UK) and 6-α-D-glucosyl-maltotriose from Megazyme (Dublin, Ireland) and were used as received and kept dried in a desiccator cabinet to avoid moistening of high DP (degree of polymerization) sugars. The standard mixture solution was composed of the 18 sugars in concentrations ranging from 0.075 to 0.25 mg.ml-1. Koji-family standard molecules [α-(12) linkages] were prepared as described in Goffin et al. (2009a). Guarantee grade reagent 50% sodium hydroxide concentrated solution used to prepare mobile phase was purchased from J. T. Baker BV (Deventer, The Netherlands). Sodium acetate trihydrate for analysis was purchased from Merck KGaA (Darmstadt, Germany). Two commercial enzymes were tested: the α-glucosidase (EC. 3.2.1.20.) L “Amano” from Amano Enzyme INC. (Nagoya, Japan) and the glucosyltransferase (EC. 2.4.1.24) L-500 from Genencor International INC. (Leiden, The Netherlands). For the enzyme activity assays, p-nitrophenyl α-D-glucoside and Trizma solution 2% w/v were purchased from Sigma-Aldrich SA (Bornem, Belgium).
2.2. AEC-PAD analysis of transglucosylation products
5The chromatography system used was an ICS-3000 system (Dionex, Sunnyvale, CA, USA). This system consisted of an SP gradient pump system, a DC detector/chromatography module thermally regulated with a 25 µL injection loop, an AS40 autosampler, and an ED40 electrochemical detector equipped with an amperometric cell. The cell comprised an about 1 mm diameter gold working electrode, a glass and Ag/AgCl combination reference electrode (Dionex) and a titanium counter electrode consisting of the cell body. The chromatographic separation of the sugars was performed on a CarboPac PA100 analytical column (250 mm × 4 mm i.d. Dionex) and a CarboPac PA10 guard column (40 mm × 4 mm i.d. Dionex) at a flow-rate of 1 mL⋅min-1; both columns were packed with an identical microporous, polymeric anion-exchange material and were installed in the thermal compartment at a controlled temperature of 30°C. The sample injection volume was 25 µl. The gradient elution was performed with mobile phases A (100 mM sodium hydroxide solution) and B (600 mM sodium acetate trihydrate in a 100 mM sodium hydroxide solution) using the gradient program described by Goffin et al. (2009a).
2.3. Enzymatic activities
6Transglucosidic activities of enzymes are determined using the method described by Megazyme with p-nitrophenyl α-D-glucoside as substrate. In standard conditions, transglucosidic activities are measured at 40°C for 10 min in a 400 µl final volume of 200 mM acetate buffer (pH 4.0) containing 10 mg of enzyme solution (with densities of 1.118 g.ml-1 and 1.17 g.ml-1 for L “Amano” and L-500 respectively) and 5 mM of p-nitrophenyl α-D-glucoside. Reaction is stopped by addition of 3 ml of a 2% w/v Trizma Base solution (pH 8.5). Absorbance due to yellow nitrophenolate ions released is read at 400 nm with a Ultraspec 4000 (Pharmacia Biotech) UV/visible spectrophotometer. The specific activity is expressed in enzymatic unit per mg of enzyme preparation (EU.mg-1). An Enzymatic Unit represents 1 µM of p-nitrophenol released per minute.
2.4. Transglucosylation reactions
7Transglucosylation enzymatic reactions take place at 60°C in 0.5 l reaction medium with 0.1 M sodium acetate buffer (4.8 ≤ pH ≤ 5.2), 0.5 ml of enzyme solution and 100 g.l-1 of maltose. For the reaction with the two enzymes in combination, 0.25 ml of each enzyme preparation is added to the buffered medium. Evolution of the synthesis reactions is monitored in time by sampling aliquots every 30 min (2 ml). Reactions are stopped by heating aliquots in a boiling water bath for 5 min. Before analysis, samples are diluted 100 times in 18 MΩ purified water.
3. Results and discussion
3.1. Detection of transglucosylation products
8The use of pure maltose as the transglucosylation reaction substrate, instead of hydrolyzed starch used in industry, leads to a simplified IMO profile (Figure 1). Moreover, the use of a step-forward AEC-PAD method (Goffin et al., 2009a) permitted us to bring into light the presence of unknown saccharides not detected with previous analytical methods. Structures and abbreviations of below discussed oligosaccharides are presented in table 1. Saccharides depicted with an asterisk (*) are present in the AEC-PAD chromatogram but their identity, structure and DP are only speculated while unmarked saccharides correspond to known compounds for which standards exist. In the chromatogram of glucosyltransferase products presented in figure 1, ten peaks correspond to known saccharides according to their retention time (RT) and the most abundant are I2, I3, M2 and P while K2, K3, N2, I4 and N3 are present in lower but significant quantities. On the other hand, at least seven other peaks before 27 min can be pointed out with 13* (22.93 min), 18* (25.4 min) and 20* (26.98 min) being the most important. The same known and unknown saccharides are present in the α-glucosidase chromatogram but sometimes in significantly different proportions (data not shown).

9Regarding the enzymatic reaction, α-glucosidase and glucosyltransferase from Aspergillus niger both catalyze the glucose transfer mainly through α-(16) linkages or in a lesser extent and in the following preferred order: α-(12), α-(13) and rarely α-(14) linkage (Kita et al., 1991; Duan et al., 1995; Vetere et al., 2000; Yamamoto et al., 2004; Barker et al., 1953; Pazur et al., 1977; 1978; Benson et al., 1982; McCleary et al., 1989). Considering these facts and starting from maltose (M2), the enzymes can thus produce trisaccharides directly: P, in lower proportions K-M2 and scarcely N-M2 while releasing a Glc residue. As Glc accumulates, disaccharides can be formed: I2, in lower proportions K2 and scarcely N2. Thus, early in the reaction, P and I2 are the main products. Later in the reaction, the produced IMO can be used as acceptors and potentially as glucosyl-donors but less efficiently than M2 (McCleary et al., 1989). Disaccharides can then be further transglucosylated to produce mainly I3, K-I2 and I-K2 while trisaccharides can be converted mainly to I-P, K-P and I-K-M2.
3.2. Endeavour of structural identification of unknown IMO
10In this section, unknown IMO structures are proposed according to their RT. Indeed, besides analysis conditions (Cataldi et al., 2000) and degree of polymerization (Li et al., 1996), relative acidities of the hydroxyl groups (Lee, 1990; Paskach et al., 1991) and their accessibility to the stationary phase functional groups (Hardy et al., 1988) are the main factors governing AEC chromatography. Oligosaccharides retention is thus driven by linkage type and spatial conformation while increasing with DP value. Thus, the presence of branching, intramolecular H bridges or the formation of suprastructures such as coil for higher DP decrease the accessibility to the –OH (Koizumi et al., 1991).
113D structures of some oligosaccharides resumed in table 1 have been constructed using SWEET II modeling program which uses MM3 force field for optimization (Figure 2). Glucose residues in oligosaccharides are named respectively A, B, C and D starting from the reducing end. Considering DP3 oligosaccharides 3D structures, relative elution position of standards can be explained by several structural characteristics. K3 and I3 are the less retained compounds (12.95 and 13.8 min) while M3 and N3 (26.29 and 27.95 min) present an accentuate retention. K3 slight retention can be explained, first, by the loss of two 2-OH which are the most acidic hydroxyl besides 1-OH. Secondly, the molecule exhibits a folded conformation stabilized by hydrogen bondings B 1-O/C 2-OH and A 1-O/C 2-OH which lower the acidity of both A 1-OH and C 2-OH and make them less accessible. Finally, the flexible and acid 6-OH are spread outside the formed “coil” giving no opportunity for an effective interacting surface. The same spreading out is observed for the 2-OH and the C 6-OH in I3 while two 6-OH are lost in the linkages. Moreover, the high flexibility of α-(1-6) linkages generates a high entropy and leads to the formation of a pseudo-coil structure which hinders the interactions with the phase (Paskach et al., 1991). On the other hand, M3 and N3 formation leads to the loss of two 4-OH and 3-OH, respectively, which are less acidic. They are both very linear molecules, thus presenting a large surface for potential interaction. In M3, the 1-OH and the three 2-OH are accessible and aligned due to the stabilizing effect of the H-bonding between 3-OH and 2-O of the next residue. The three 6-OH are also accessible and aligned on the other side of the molecule. In N3, 2-OH and 6-OH are both accessible and are alternating on both sides every residue. Furthermore, considering P (23.55 min) and I-M3 (29.65 min), these IMO both possess α-(1-4) linkages and a single α-(1-6) at the non-reducing end. The presence of this linkage reduces the retention time compared to M3 (26.29 min) and M4 (31.74 min), because it alters the linearity of the molecule and moreover, an H-bonding can be formed between A 6-OH and C 2-OH. It is worth noting that K4, I4, K5 and I5 are all eluted before P demonstrating the importance of the overall structure on retention.
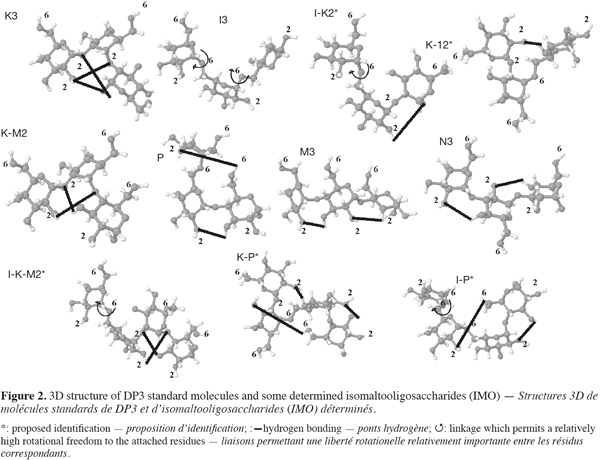
12Peaks 6* (15.6 min) and 10* (19.7 min) are eluted after K3, I3 and before I4, P, M3 and N3 and both present a low area. This suggests that they are both DP3 presenting α-(1-2) and α-(1-6) linkages as they are present in significant quantity and corresponding therefore to I-K2* or K-I2*. I-K2* possess a flexible α-(1-6) linkage which makes the C-residue relatively mobile while the same linkage in K-I2* is stabilized by the presence of a H-bond between C 2-O and A 4-OH. Moreover, A 1-OH and B 2-OH acidities in I-K2* are reduced due to a potential H-bond formation between these hydroxyls. In K-I2*, A 1-OH and A 2-OH are accessible as well as B and C 6-OH. Peak 6* and 10* are thus more likely to correspond to I-K2* and K-I2*, respectively. Peak 13* (22.93 min) present a relatively high area and is eluted after K4 and I4 and just before P and M3. It should thus correspond to a DP3 with a “low retention” linkage [α-(1-2) or α-(1-6)] and a “high retention” linkage [α-(1-3) or α-(1-4)]. The peak’s high area suggests that the linkage formed quite easily by the enzyme giving no chance to α-(1-3) and α-(1-4) at the non reducing end. K-M2* known as centose is thus the best candidate as maltose is the most abundant acceptor during most of the transglucosylation process. Moreover, the presence of an H-bond between C 2-O and A 3-OH reduces the accessibility to A 1-OH and the 2-OH in comparison to P. Peaks 16* (24.2 min), 18* (25.4 min) and 20* (26.98 min) are all eluted after P and before N3 and I-M3, and 20* is co-eluted with M3 (data not shown). They are thus likely to be DP4 with mixed linkages and products of DP3 transglucosylation. The most abundant DP3 and in order of importance are P, I3 and speculated K-M2*. K-I3* is susceptible to have a short retention time and can, thus, correspond to the small peak 11’ while I4 retention time is known (20.4 min). Other DP4 peaks can thus correspond, and in order of importance, to I-P*, K-P* and I-K-M2*. According to abundance, peaks 16*, 18* and 20* could correspond to I-K-M2*, K-P* and I-P*, respectively. When looking at 3D structures, I-K-M2 has a compact and similar structure as K-M2* with a non-reducing residue linked by a flexible α-(1-6). Its retention time should therefore be shorter than those of K-P and IP in the same way as for K-M2* and P. When comparing I-P* and K-P*, the first exhibits a more linear structure while the second shows restricted accessibility due to the presence of several H-bonds.
3.3. Study of the reaction products of two types of transglucosylating enzymes on maltose
13Two commercial enzymes, L-500, a glucosyltransferase (EC. 2.4.1.24), and L-AMANO, an α-glucosidase (EC. 3.2.1.20), were tested alone and in combination on pure maltose for their transglucosylation specificity. Their activities were, respectively, 3.79 and 4.52 EU.mg-1 of commercial preparation. The evolution of the different products (except maltose) is presented in figure 3. Indeed, saccharides can successively be produced, increasing their relative abundance or used as donor (hydrolyzed) and as acceptor, decreasing their relative abundance.
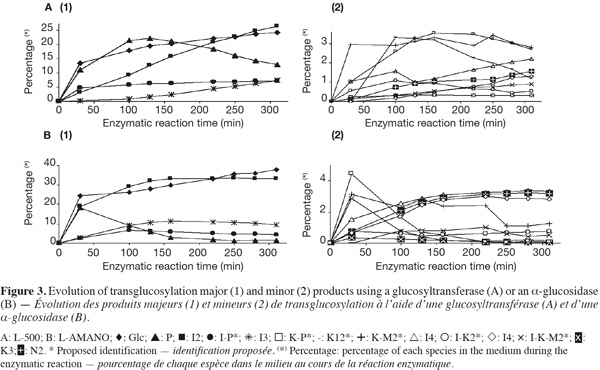
14Only IMO presented in table 1 and corresponding to standards or unknown IMO described in section 3.2. and inside the LOQ are considered. However, other oligosaccharides of DP ≥ 4 only represent 2 to 3% of the total oligosaccharides, as transglucosidase from Aspergillus niger only acts on oligosaccharides with a low degree of polymerization (Pazur et al., 1978; Benson et al., 1982; McCleary et al., 1989). Areas were corrected according to response factors determined from the standard molecules calibration curves. Theoretical response factors of other oligosaccharides were calculated according to their retention times and as a consequence to their DP and linkage types (data not shown). Indeed, as retention times, response factors are linked to the oligosaccharide structure by their relative acidity and accessibility of their hydroxyl group and moreover, their diffusion coefficient in the electrode. Concerning the most abundant oligosaccharides [Figure 3(1)], not surprisingly, the α-glucosidase (L-AMANO) has a stronger hydrolyzing activity and works much faster. Indeed, after only 30 min, it produces similar and high amounts of P and I2, even more Glc and some I3 while the glucosyltransferase (L-500) produces less P, Glc and low amounts of I2 and I3 compared to the α-glucosidase. Then L-AMANO rapidly hydrolyzes P and produces more I2 and I3 while L-500 continues to produce P and increasing amounts of I2 and I3. After 150 min, L-AMANO only produces more Glc while I2 and I3 contents stay relatively constant indicating that the enzyme easily hydrolyzes α-(1-4) linkages of maltose but also α-(1-6) linkages presenting a neighboring α-(1-4) such as in P. For L-500, P only starts to be hydrolyzed. At the end of the reaction, I2 and I3 contents were significantly higher for L-AMANO than for L-500.
15Similar observations can be done for minor products [Figure 3(2)]. For L-AMANO, small amounts of three unknown IMO speculated as I-K2*, I-K-M2*, K-I2*, respectively and K3 and N3 are produced while approximately 3% of I4, N2 and K2 are produced and not hydrolyzed. On the other hand, relatively high amount of two other unknown IMO speculated as K-P* (4.47%) and K-M2* (3.16%) are produced but easily hydrolyzed especially K-P* as for P. For L-500, products are again less easily hydrolyzed except for speculated K-I2*. Indeed, speculated K-M2* and K-P* are still present at, respectively, 2.82 and 2.70% after 310 min. As a general rule, the α-glucosidase has a more expressed hydrolyzing activity leading to products containing less residual digestible α-(1-4) linkages such as I2, I3, I4, K2 and N2 while the glucosyltransferase produces important amounts of P, I2 and speculated K-M2*, I-P* and K-P*. IMO profiles can, thus, be orientated through the enzyme choice.
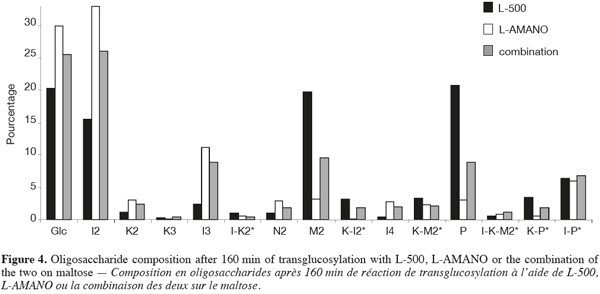
16When using the two enzymes in combination (Figure 4), after 160 min an intermediate profile is obtained, each product being present in percentages in between the values obtain for the enzymes alone. Concerning the IMO content after 160 min, 66.78, 59.86 and 64.81% of IMO are found for L-AMANO, L-500 and their combination, respectively. Thus the enzymes do not seem to show a synergistic effect neither an inhibiting effect on each other. However, similar maximum IMO contents of 67.65 and 66.62% are obtained after 130 and 310 min for L-AMANO and L-500, respectively, even though, IMO preparations produced using L-AMANO contain less digestible IMO as they present less residual α-(1-4) linkages. This confirms the fact that L-AMANO is working much faster and reactions must be stopped on time to avoid hydrolysis of produced IMO. Moreover, after 160 min or at the maximum IMO content (130 min for L-AMANO and 310 min for L-500), L-AMANO presents a higher content in Glc and a lower content of M2 compared to L-500. This is worth noting because Glc is easier to eliminate from the IMO preparation by yeast fermentation (Pan et al., 2005) or conversion to prebiotic gluconic acid (Goffin et al., 2009b) than M2 in order to obtain better prebiotic index.
4. Conclusion
17In this paper, transglucosylation reaction leading to IMO production has been studied on pure maltose using two types of enzymes: a glucosyltransferase (EC. 2.4.1.24) and an α-glucosidase (EC. 3.2.1.20). The reactions were monitored using a step-forward AEC-PAD analytical method which permitted to detect and resolve new unknown IMO. Structural determination of unknown IMO was attempted using their RT and relative abundance in relation with the enzyme propensity to form different linkages. Two different IMO profiles were found for the two enzymes demonstrating the importance of the enzyme specificity on the final product. As a general rule, the α-glucosidase has a more expressed hydrolyzing activity leading to products containing less residual digestible α-(1-4) linkages such as I2, I3, I4, K2 and N2 while the glucosyltransferase produces important amounts of P and speculated unknown IMO K-M2*, I-P* and K-P* respectively. Finally, the combination of the two enzymes leaded to an intermediate IMO profile. Consequently, this paper permitted us, first, to detect the presence of unidentified IMO thanks to the use of a step-forward chromatographic method and thereafter proposing possible structure in relation to their retention times and abundance. Moreover, IMO syrups composition was proved to be dependant on the specificity of the transglucosylating enzyme so that products profiles can be therefore designed using different enzymes and in different proportion.
18Abbreviations
19ABEE: p-aminobenzoic ethyl ester
20AEC-PAD: Anion exchange chromatography-Pulsed amperometry detection
21ALO: Anomalously linked oligosaccharides
22IMO: isomaltooligosaccharides
23DP: degree of polymerization
24RT: retention times
25LOQ: limits of quantification
26EU: enzymatic units
273D: 3 dimensions
28NMR: nuclear magnetic resonance
29LC-MS: liquid chromatography – mass spectrometry
30Acknowledgements
31This work was jointly supported by the Ministry of the Walloon Region and the company Meurens Natural throughout the FIRST DEI IMOBIOSE research project.
Bibliographie
Ammeraal R.N., Delgado G.A., Tenbarge F.L. & Friedman R.B., 1991. High-performance anion-exchange chromatography with pulsed amperometric detection of linear and branched glucose oligosaccharides. Carbohydr. Res., 215, 179-192.
Barker S.A. & Carrington T.R., 1953. Studies of Aspergillus niger. Part II. Transglycosidation by Aspergillus niger. J. Chem. Soc. London, 3588-3593.
Benson C.P., Kelly C.T. & Fogarty W.M., 1982. Production and quantification of transglucosidase from Aspergillus niger. J. Chem. Technol. Biotechnol., 32, 790-798.
Cataldi T.R.I., Campa C. & De Benedetto G.E., 2000. Carbohydrate analysis by high-performance anion-exchange chromatography with pulsed amperometric detection: the potential is still growing. Fresenius J. Anal. Chem., 368, 739-758.
Chiba S., 1997. Molecular mechanism in α-glucosidase and glucoamylase. Biosci. Biotechnol. Biochem., 61, 1233-1239.
Demuth K., Jordening H.J. & Buchholz K., 2002. Oligosaccharide synthesis by dextransucrase: new unconventional acceptors. Carbohydr. Res., 337, 1811-1820.
Dols-Lafargue M., Willemot R.M., Monsan P.F. & Remaud-Simeon M., 2001. Factors affecting alpha-(1-2) glucooligosaccharide synthesis by Leuconostoc mesenteroides NRRL B-1299 dextransucrase. Biotechnol. Bioeng., 74(6), 498-504.
Duan K.J., Sheu D.C., Lin M.T. & Hsueh H.C., 1994. Reaction mechanism of isomaltooligosaccharides synthesis by α-glucosidase from Aspergillus carbonarious. Biotechnol. Lett., 16(11), 1151-1156.
Duan K.J., Sheu D.C. & Lin C.T., 1995. Transglucosylation of a fungal α-glucosidase. The enzyme properties and correlation of isomaltooligosaccharide production. Ann. N.Y. Acad. Sci., 750, 325-328.
Faijes M. & Planas A., 2007. In vitro synthesis of artificial polysaccharides by glycosidases and glycosynthases. Carbohydr. Res., 342, 1581-1594.
Fernandez-Arrojo L. et al., 2007. Transformation of maltose into prebiotic isomaltooligosaccharides by a novel a-glucosidase from Xantophyllomyces dendrorhous. Process Biochem., 42, 1530-1536.
Gibson G.R. & Roberfroid M.B., 1995. Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J. Nutr., 125, 1401-1412.
Goffin D. et al., 2009a. A step-forward method of quantitative analysis of enzymatically produced isomaltooligosaccharide preparations by AEC-PAD. Chromatographia, 69, 287-293.
Goffin D., Paquot M., Blecker C. & Robert C., 2009b. Process for the production of a composition, the composition and the use there-of as food additive. European patent n°2010/081913, 22/07/2010.
Goffin D. et al., 2010. Will isomaltooligosaccharides, a well established functional food in Asia, break through the European and American market : the status of knowledge on these prebiotics. Crit. Rev. Food Sci. Nutr., accepted.
Gu Q., Yang Y., Jiang G. & Chang G., 2003. Study on the regulative effect of isomaltooligosaccharides on human intestinal flora. J. Hyg. Res., 32, 54-55.
Hamada S. & Torii M., 1980. Interaction of glucosyltransferase from S. mutans with various glucans. J. Gen. Microbiol., 116, 51-59.
Hardy M.R. & Townsend R.R., 1988. Separation of positional isomers of oligosaccharides and glycopeptides by high-performance anion-exchange chromatography with pulsed amperometric detection. Proc. Natl Acad. Sci. USA, 85(10), 3289-3293.
Kaneko T. et al., 1994. Effects of isomaltooligosaccharides with different degrees of polymerization on human fecal bifidobacteria. Biosci. Biotechnol. Biochem., 58(12), 2288-2290.
Kaneko T. et al., 1995. Evaluation of acidogenicity of commercial isomaltooligosaccharides mixture and its hydrogenated derivative by measurement of pH responses under human dental plaque. Biosci. Biotechnol. Biochem., 59, 372-377.
Kanno T., 1990. Some functional properties of so-called isomaltooligosaccharides and their applications to food industry. Denpun Kagaku, 37, 87-97.
Kato N. et al., 2002. Novel α-glucosidase from Aspergillus nidulans with strong transglycosylation activity. Appl. Environ. Microbiol., 68, 1250-1256.
Kita A. et al., 1991. Substrate specificity and subsite affinities of crystalline α-glucosidase from Aspergillus niger. Agric. Biol. Chem., 55, 2327-2335.
Kobayashi I. et al., 2003. Purification and characterization of a new type of α-glucosidase from Paecilomyces lilacinus that has transglucosylation activity to produce α-(1,3)- and α-(1,2)-linked oligosaccharides. Biosci. Biotechnol. Biochem., 67, 29-35.
Kohmoto T. et al., 1988. Effect of isomaltooligosaccharides on human fecal flora. Bifidobacteria Microflora, 7, 61-69.
Kohmoto T., Fukui F., Takaku H. & Mitsuoka T., 1991. Dose-response test of isomaltooligosaccharides for increasing fecal bifidobacteria. Agric. Biol. Chem., 55(8), 2157-2159.
Koizumi K., Kubota Y., Tanimoto T. & Fukuda M., 1989. High performance anion-exchange chromatography of homogeneous D-gluco-oligosaccharides and poly-saccharides (polymerization degree > 50) with pulsed amperometric detection. J. Chromatogr., 464, 365-373.
Koizumi K. & Fukuda M., 1991. Estimation of the distributions of chain length of amylopectins by high-performance liquid chromatography with pulsed amperometric detection. J. Chromatogr., 585, 233-238.
Lee Y.C., 1990. High-performance anion-exchange chromatography for carbohydrate analysis. Anal. Biochem., 189, 151-162.
Li Z., Mou S., Liao W. & Lu D., 1996. The study of the relationship between retention and structure on D-mannose and its derivatives with high-performance anion-exchange chromatography. Carbohydr. Res., 295, 229-234.
McCleary B. & Gibson T., 1989. Purification, properties, and industrial significance of transglucosidase from Aspergillus niger. Carbohydr. Res., 185, 147-162.
Nakanishi T., Nomura S. & Takeda Y., 2006. An improved method for the quantitative analysis of commercial isomaltooligosaccharides products using the calibration curve of standard reagents. J. Appl. Glycosci., 53, 215-222.
Nishi K., Chiba S. & Shimomura T., 1975. Enzymatic synthesis of a branched trisaccharide, 2,4-di-α-glucosyl-glucose. Agric. Biol. Chem., 39(3), 727-728.
Pan Y.-C. & Lee W.-C., 2005. Production of high-purity isomaltooligosaccharides syrup by the enzymatic conversion of transglucosidase and fermentation of yeast cells. Biotechnol. Bioeng., 89(7), 797-804.
Paskach T., Licker H.P., Reilly P.J. & Thielecke K., 1991. High-performance anion-exchange chromatography of sugars and sugar alcohols on quaternary ammonium resins under alkaline conditions. Carbohydr. Res., 215, 1-14.
Pazur J.H., Cepure A., Okada S. & Forsberg L.S., 1977. Comparison of the action of glucoamylase and glucosyltransferase on D-glucose, maltose, and maltooligosaccharides. Carbohydr. Res., 58, 193-202.
Pazur J.H., Tominaga Y., DeBrosse C.W. & Jackman L.M., 1978. The synthesis of 1,6-anhydro-β- -glucopyranose and -glucosyl oligosaccharides from maltose by a fungal glucosyltransferase. Carbohydr. Res., 61, 279-290.
Perugino G., Trincone A., Rossi M. & Moracci M., 2004. Oligosaccharide synthesis by glycosynthases. Trends Biotechnol., 22(1), 31-37.
Plou F.J. et al., 2002. Glucosyltransferases acting on starch or sucrose for the synthesis of oligosaccharides. Can. J. Chem., 80, 743-752.
Plou F.J., Gomez de Segura A. & Ballesteros A., 2007. Application of glycosidases and transglycosidases in the synthesis of oligosaccharides. In: Polaina J. & MacCabe A.P., eds. Industrial Enzymes. Dordrecht, The Netherlands: Springer, 141-157.
Roberfroid M.B., 2008. Prebiotics: concept, definition, criteria, methodologies, and products. In: Gibson G.R. & Roberfroid M.B., eds. Handbook of prebiotics. Boca Raton, FL, USA: CRC Press, Taylor and Francis, 40-60.
Rycroft C., Jones M., Gibson G. & Rastall R., 2001. The role of prebiotics in human gut microbiology. Prebiotic oligosaccharides. J. Appl. Microbiol., 91, 878-887.
Sanz M.L., Gibson G.R. & Rastall R.A., 2005. Influence of disaccharide structure on prebiotic selectivity in vitro. J. Agric. Food Chem., 53(13), 5192-5199.
Takahashi S. & Nagayama K., 1988. A novel NMR microcell with symmetric geometry. J. Magn. Reson., 76, 347-351.
Takaku H., 1988. Anomalously linked oligosaccharides mixture (“Alo mixture”). In: The Amylase Research Society of Japan, ed. Handbook of amylase and relative enzymes. Oxford , UK: Pergamon Press, 215-217.
Vetere A., Gamini A., Campa C. & Paoletti S., 2000. Regiospecific transglycolytic synthesis and structural characterization of 6-O-a-glucopyranosyl-glucopyranose (isomaltose). Biochem. Biophys. Res. Commun., 274, 99-104.
Vinogradov E. & Bock K., 1998. Structural determination of some new oligosaccharides and analysis of the branching pattern of isomaltooligosaccharides from beer. Carbohydr. Res., 309, 57-64.
Yamamoto T. et al., 2004. Purification and characterization of Acremonium implicatum a-glucosidase having regioselectivity for α-(1,3)-glucosidic linkage. Biochim. Biophys. Acta, 1700, 189-198.
Wang Y., 2009. Prebiotics: present and future in food science and technology. Food Res. Int., 42, 8-12.
Yun J., Suh J.H. & Song S., 1994. Kinetic study and mathematical model for the production of isomalto-oligosaccharides from maltose by transglucosylation of Aureobasidium pullulans. J. Korean Inst. Chem. Eng., 32(6), 875-880.
Pour citer cet article
A propos de : Dorothée Goffin
Univ. Liege - Gembloux Agro-Bio Tech. Department of Industrial Biological Chemistry. Passage des Déportés, 2. B-5030 Gembloux (Belgium). E-mail: dorothee.goffin@ulg.ac.be – Univ. Liege - Gembloux Agro-Bio Tech. Department of Food Technology. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
A propos de : Bernard Wathelet
Univ. Liege - Gembloux Agro-Bio Tech. Department of Industrial Biological Chemistry. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
A propos de : Christophe Blecker
Univ. Liege - Gembloux Agro-Bio Tech. Department of Food Technology. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
A propos de : Claude Deroanne
Univ. Liege - Gembloux Agro-Bio Tech. Department of Food Technology. Passage des Déportés, 2. B-5030 Gembloux (Belgium).
A propos de : Yves Malmendier
Meurens Natural. Rue des Martyrs, 21. B-4650 Herve (Belgium).
A propos de : Michel Paquot
Univ. Liege - Gembloux Agro-Bio Tech. Department of Industrial Biological Chemistry. Passage des Déportés, 2. B-5030 Gembloux (Belgium).