


- Accueil
- Volume 21 (2017)
- Numéro 1
- The influence of amplicon length on real-time PCR results


Visualisation(s): 13264 (77 ULiège)
Téléchargement(s): 1693 (8 ULiège)

The influence of amplicon length on real-time PCR results

Notes de la rédaction
Received on July 15, 2016; accepted on January 23, 2017
This article is distributed under the terms and conditions of the CC-BY License (http://creativecommons.org/licenses/by/4.0)
Résumé
Influence de la taille de l’amplicon sur les résultats obtenus par PCR en temps réel
Description du sujet. Cet article traite de l’influence de la taille de l’amplicon sur les résultats obtenus par PCR en temps réel.
Objectifs. Le but des expériences menées vise à montrer que la taille de la région ciblée a une influence sur la détection.
Méthode. Les expériences ont été réalisées sur de l’ADN génomique et plasmidique. Les techniques d’amplification par PCR en temps réel ont été effectuées soit au moyen de sondes d’hybridation ou de SYBR® Green. Des amorces ont été sélectionnées de manière à produire des fragments de tailles croissantes. Les expériences ont été réalisées sur deux cibles : une cible endogène au soja (portion du gène de la lectine) et une cible transgénique (jonction P35S-CTP de la construction du soja MON40-3-2).
Résultats. Les résultats ont montré que la cinétique d’amplification évolue en fonction de la taille de l’amplicon et que les plus petits amplicons atteignent un plus haut niveau de fluorescence en phase plateau. L’utilisation d’un ADN dégradé ainsi que la chimie utilisée ont un impact sur les résultats.
Conclusions. Il a été démontré expérimentalement que l’effet observé est lié à la température d’élongation suboptimale utilisée en PCR en temps réel. Le format de PCR utilisant le SYBR® Green est cependant moins impacté car la perte d’efficience est partiellement compensée par l’intégration d’un nombre de molécules de SYBR® Green plus important dans les plus grands fragments.
Abstract
Description of the subject. This paper discusses the influence of amplicon length on real-time PCR results.
Objectives. The aim of the experiments was to show that amplicon size has an influence on detection.
Method. Tests were performed on genomic and plasmid DNA. Double-dye probes and SYBR® Green were used for detection by real-time PCR. Primers were selected in order to produce fragments with increasing sizes. Experiments dealt with two targets: an endogenous target for soybean (part of the lectin gene) and a transgenic target (junction P35S-CTP of the MON40-3-2 soybean).
Results. The results show that the kinetics of amplification curves evolve as a function of amplicon length, and smaller amplicons yield a higher level of fluorescence for the plateau phase. DNA degradation within the sample as well as the principles of fluorescence acquisition as a function of the chemistry used can also be factors.
Conclusions. It was experimentally shown that the observed effect is linked to the suboptimal elongation temperature used in real-time PCR. Detection using SYBR® Green is less impacted as the loss of efficiency is partially compensated by the greater integration of SYBR® Green molecules in the larger fragments.
Table des matières
1. Introduction
1Genetic modified organisms detection in agrifood products can be subject to difficulties due to food processing techniques, inducing DNA degradation and reduction into smaller DNA pieces (Meyer et al., 1996; Gryson et al., 2010). In processed products, DNA integrity is a limiting factor for extraction. Problems may also arise during DNA amplification. Inhibitors may for instance hinder the progress of amplification (Kontanis & Reed, 2006; Opel et al., 2010). Therefore the choice of appropriate primers is a determining factor.
2Couples of primers may present different efficiencies and the formation of primer-dimers (Miller et al., 1996) must be minimized during primer selection, using programs providing support for primer design. The formulation of reagents is another factor that can affect the sensitivity of the reactions (Farriol & Orta, 2005).
3Most companies providing tools or reagents (Applied Biosystems, 2005; Bio-rad Laboratories, 2006; Qiagen, 2011) state that amplicon size plays a role in amplification efficiency by real-time PCR. They generally recommend the use of relatively short amplicon lengths, in the range of 50 to 150 base pairs (bp). To the best of our knowledge, no study has clearly demonstrated such an influence using real-time PCR chemistries.
4The use of real-time PCR also makes it possible to amplify targets with a size below 100 bp. Such amplifications are more difficult with classical PCR systems due to the fact that such small fragments are less easy to visualize on an agarose gel of the kind used in classical PCR. Such visualization is possible by the integration of ethidium bromide molecules between the two DNA strands, which is evidenced by the fluorescence emitted under UV exposure. As the integration of ethidium bromide is directly proportional to the DNA fragment length, the longer the fragments, the more visible they are. In real-time PCR too, chemistries used for DNA detection may also have an impact on sensitivity. There are two main chemistries used: intercalating agents and the double-dye (TaqMan®) probes format.
5– SYBR® Green format: SYBR® Green, like ethidium bromide, is a molecule with an affinity for double-stranded DNA; its fluorescence is largely enhanced once it is bound to the major groove of DNA (Higuchi et al., 1993). SYBR® Green is used in real-time PCR to monitor amplification kinetics, because the fluorescence increases in proportion to the number of amplicons (Ririe et al., 1997). During the denaturation step, all SYBR® Green molecules intercalated between the two strands are released and used again at the next amplification cycle (Figure 1). Technical data sheets claim that SYBR® Green can be approximately 25-100 times more sensitive than ethidium bromide staining (Invitrogen, 2006).

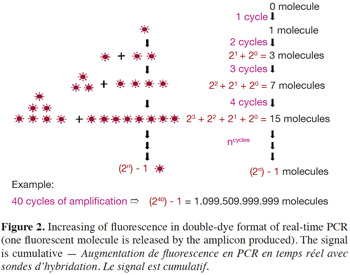
6– Double-dye probes: in real-time PCR with double-dye probes like TaqMan® probes, detection is performed through fluorescence emitted by the dyes released due to the exonuclease activity of Taq polymerase (Holland et al., 1991). Double-dye probes cannot be used again once they have been degraded by exonuclease activity of the Taq polymerase. The fluorescence produced by the cleavage of new probes is added to fluorescence acquired during previous cycles (Figure 2). Fluorescence is thus not a direct measurement of the number of amplicons of the last cycle, but cumulatively reflects the fluorescence generated during all previous cycles.
7The sensitivity of detection by a well-designed real-time PCR test in comparison with a classical PCR test can be explained by the fact that fluorescence is measured by a CCD (charge coupled device) camera and is more sensitive than end-point detection PCR with agarose gel electrophoresis. The second explanation is linked to the size of the amplified fragments. Targets in real-time PCR can be very short and generally do not exceed 120 bp. This is very useful for detecting the target when DNA is degraded. In classical PCR, fragments are generally longer to ensure easier visualization on agarose gel.
8In this study the influence of amplicon length on PCR yield in real-time PCR was analyzed, to show that the advantage of using small targets for GMO detection is linked to an intrinsic real-time PCR property.
2. Materials and methods
9The samples used were commercial feed samples and certified reference materials (CRMs). The CRMs were developed by the Institute for Reference Materials and Measurements (IRMM, Geel, Belgium) and consisted of dried soybean powder (IRMM 410) containing mass fractions at 5%, 2%, 1% and 0.1% of Roundup Ready soybean (event MON40-3-2).
10Assays were also performed on plasmid DNA. The plasmid pl-RR used contains a 528 bp fragment issued from the Roundup Ready construction. This fragment was inserted in pCRII plasmid with the TOPO TA cloning kit (Invitrogen, Merelbeke, Belgium). The pl-Lec plasmid was built at CRA-W in a similar way and contains a 501 bp fragment of the lectin gene.
11DNA isolation on feed and CRMs was performed with the Wizard Magnetic DNA Purification System for Food kit (Promega, Madison, USA) on a test portion of 50 mg. DNA was eluted in 150 µl of elution buffer. Extraction was performed on the King Fisher robot (Thermo Fisher, Hampshire, UK). Between 10 and 100 ng were used in the PCR tests, corresponding to the quantity of DNA generally introduced in PCR tubes for routine analysis. The quantities used may therefore be different from one sample to another. However, the quantities used are always identical for the same sample, making it possible to compare Cq values in order to assess the effect of amplicon size.
12Plasmid DNA was isolated from bacterial cultures with the High Pure Plasmid Isolation kit from Roche Diagnostics (Mannheim, Germany). Plasmids were linearized with BamHI enzyme (Roche Diagnostics, Mannheim, Germany) and products of linearization were purified with the last steps of a Phenol-Chloroform extraction (Sambrook et al., 1989).
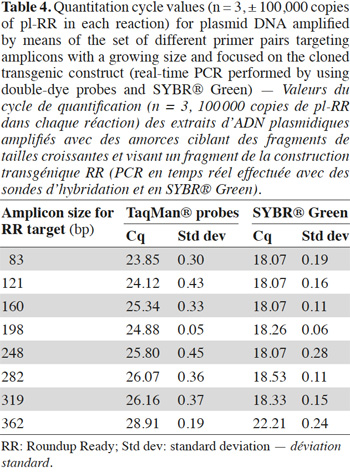
2.1. Oligonucleotides
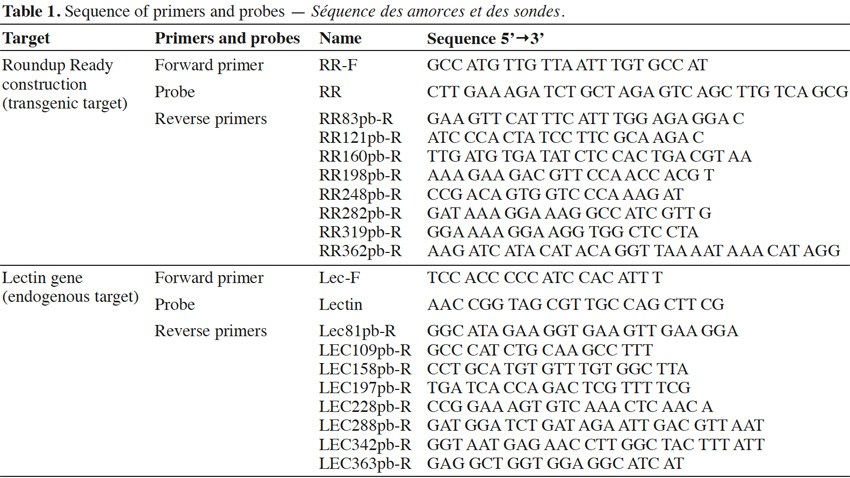
13Primers were synthesized by Invitrogen (Merelbeke, Belgium). Probes were synthesized by Eurogentec (Seraing, Belgium). The probes were labeled with the reporter dye FAM at the 5’ end and the quencher dye TAMRA at the 3’ end. Primers were chosen with Primer Express program v2.0 (Life Technologies, Foster City, CA). Primer and probe sequences are presented in table 1.
2.2. PCR conditions.
14Real-time PCR reactions (35 µl) were performed in triplicate on ABI5700 and ABI7000 PCR machines (Life Technologies, Foster City, CA) with the qPCR Mastermix (Eurogentec, Seraing, Belgium). At 17.5 µl of Master Mix, we added 2.5 µl of each primers (5 µM), 2.2 µl of probe (9 µM), 5.3 µl of bidistilled water and 5 µl of DNA.
15Reaction mixes were distributed in a 96-well reaction plate (Eurogentec). The wells were covered by optical caps (Eurogentec). The plates were centrifuged (500 rpm, 10 s) to eliminate any air bubbles at the bottom of the wells.
16PCR conditions were as follows: 2 min at 50 °C, 10 min at 95 °C and 50 cycles of 15 s at 95 °C and 1 min at 60 °C. At the end of the run, the baseline (the normalized fluorescent signal before exponential PCR amplification occurs) was adjusted and a threshold level of fluorescence was fixed in the middle of the linear region of the amplification curve represented in a semi-logarithmic graph (Y-axis with log fluorescence level as a function of cycles). This allowed the Cq values (Quantitation Cycle, sometimes also referred to as the Ct value, for Cycle threshold) of the different samples to be compared.
2.3. Targets
17Two target types are generally searched for in GMO detection: a fragment of the transgenic element inserted in the plant (NCBI accession AB209952) and an endogenous target specific to the tested plant.
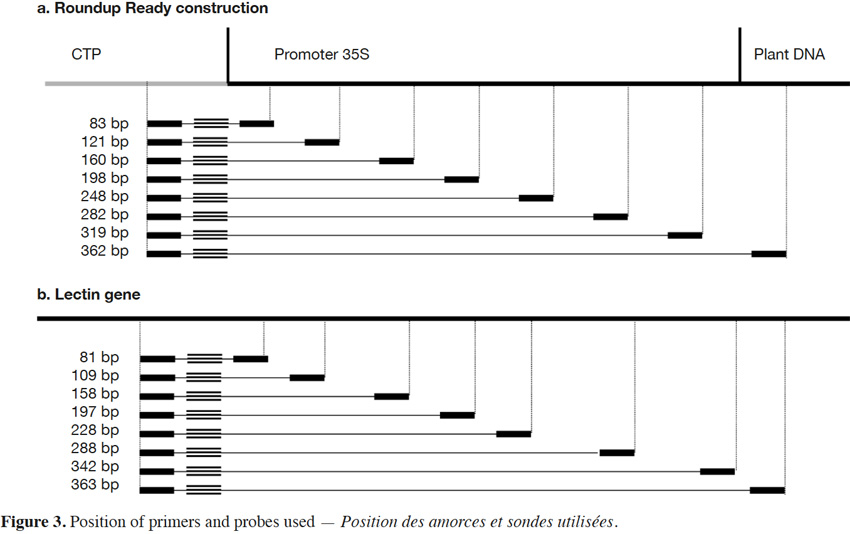
18The primers and probe used here for Roundup Ready soybean (RR) detection target the P35S-CTP junction (Bruderer & Leitner, 2003) and amplify a fragment of 83 bp. The primers were developed in order to progressively increase the amplicon size. The forward primer was conserved and the reverse primers were moved in steps of +/- 40 bp in order to increase the amplicon size. The forward primer was kept close to the probe to allow the probe to be rapidly degraded by the exonuclease activity of the Taq polymerase during the elongation step. The amplicons produced had sizes of 83, 121, 160, 198, 248, 319 and 362 bp for P35S-CTP junction (Figure 3a).
19The endogenous target commonly used for soybean detection is the lectin gene (NCBI accession K00821). Here too, the amplicons were progressively increased to reach the following sizes: 81, 109, 158, 197, 228, 288, 342 and 363 bp (Figure 3b).
2.4. DNA extracts
20DNA extracts originate from different sources:
21– Certified reference material (CRM) containing determined levels of Roundup Ready material. Soybean flours containing 1 and 5% RR soybean were tested with RR and lectin primers;
22– DNA extracts of CRM at 2% of Roundup Ready soybean were physically degraded by sonication in a previous experiment (Debode et al., 2007) and tested with the primers and probes. This treatment led to a cleavage of DNA strands and a distribution in favor of small fragments;
23– A commercial feed ingredient: a soybean oilcake, which is a very common matrix in routine GMO analyses. Kernels of an organic soybean were also used.
24– Plasmid DNA was also amplified. This was considered to be like a pure nucleic acid material for which amplifications were deemed not to be subject to a matrix effect. Two different plasmids were used. The first plasmid, pl-Lec, contains a 501 bp fragment of lectin gene and the second plasmid, pl-RR, contains a 528 bp fragment of Roundup Ready construction including left border plant-P35S junction (Windels et al., 2001) up to the P35S-CTP junction. Sequences of the largest fragments amplified with primers from this study were inside the cloned fragments.
25Experiments were performed in order to evaluate size influence on GMO detection by real-time PCR. In order to compare the results, all amplifications for lectin or RR targets were performed simultaneously for a determined matrix. Comparison between the two chemistries was performed on identical quantities of DNA per sample and with the data from a same run. This was done by selecting the appropriate filters on real-time PCR apparatus and applying a dissociation protocol at the end of the run. This had no effect on PCR amplification with double-dye probes but made it possible to observe dissociation curves for amplifications performed with SYBR® Green format. Cq values were determined by fixing a threshold level within the logarithmic part of the amplification curves. Means of Cq values are presented in the different tables.
3. Results
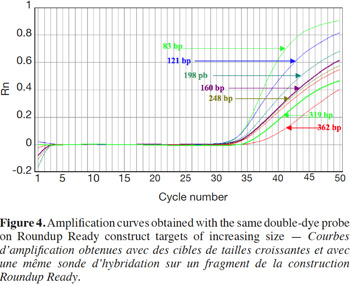
26In real-time PCR with TaqMan® probes, the amplicon size directly influenced detection: the larger the amplicons, the later the detection (Tables 2 and 3).
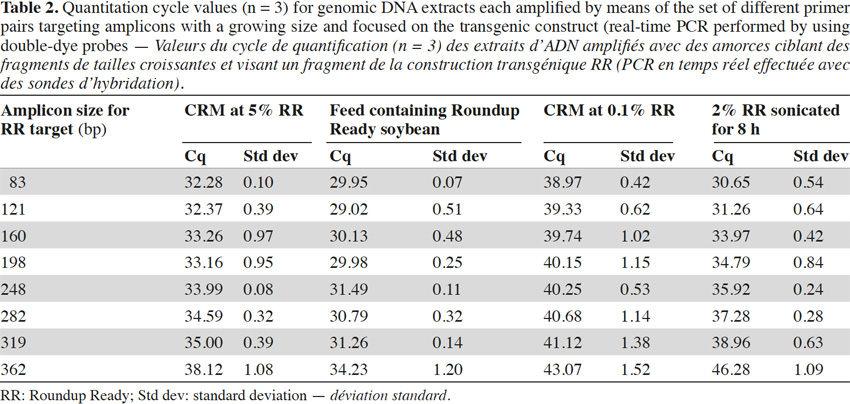
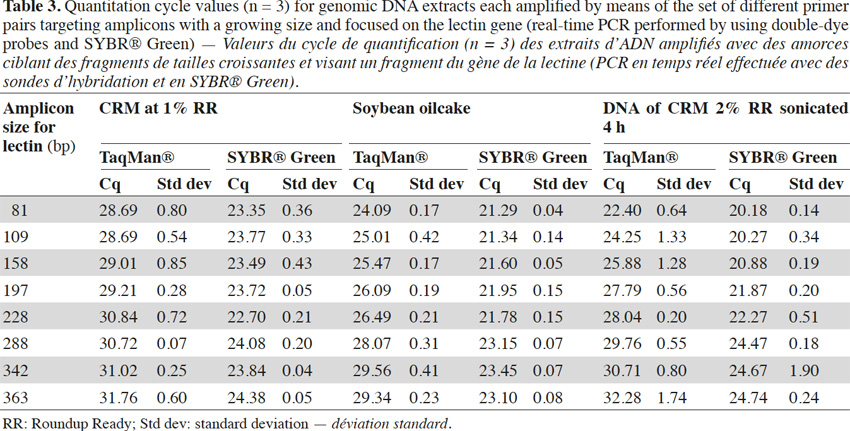
27Amplification curves of the P35S-CTP junction region for 5% RR CRM obtained with the different primer pairs are presented in linear mode in figure 3. Earlier detection (lower Cq value) and a higher fluorescence level (plateau phase) were generally observed for shorter amplicons (Figure 4). Another observation was the loss of efficiency when larger amplicons were targeted. The efficiency of amplification was evaluated through the raw data of fluorescence obtained for the construction of the amplification curves (data accessible on the ABI7000 real-time PCR device) and calculated for three different samples: DNA from reference flour at 0.1% RR, DNA from a commercial feed sample containing RR soybean and 8 h sonicated DNA from a reference flour at 2% RR.
28The efficiency of amplification was around 100% for the targets of 83 and 121 bp, and as the amplicons were elongated, efficiency showed a decreasing trend, reaching around 70% for the 0.1% RR and the commercial feed and as low as 50% for the sonicated DNA (values given for the target at 362 bp).
29These results also show that the use of larger fragments could impact the limit of detection and that targets present at low levels could not be detected due to the loss of efficiency.
30Despite modifications of the reaction mix (e.g. increases in the concentration of nucleotides, primers or probes), no improvement was obtained (not shown). For one of the targets (RR target) however, the 198 bp fragment was sometimes detected earlier than the 160 bp target. This was probably due to a better combination of primers for this 198 bp target.
31Experiments were also performed on the endogenous target for soybean, the lectin gene. Primers were selected according to Primer Express software requirements, so it was not always possible to change the position of reverse primers by exact steps of 40 bp. Experiments were performed with probes and SYBR® Green and similar conclusions to those obtained with P35S-CTP targets can be drawn when considering the results of the real-time PCR with TaqMan® probes (Table 3). With SYBR® Green the effect of a later detection of larger amplicons can be observed too, but is again less pronounced (Table 3).
32Results for CRM at 1% RR, soybean oilcake and 4 h sonicated DNA with the lectin targets are presented in table 3.
33The matrix effect is clearly visible on DNA extracts sonicated for 4 or 8 h (Tables 2 and 3). The Cq shift between the 83 bp and the 362 bp amplified fragment was about 11 cycles for a 4 h sonicated sample, and about 15.5 cycles for an 8 h sonicated DNA.
34The more the sample is damaged, the greater the influence of an increase of Cq value on large targets. This is linked to a less abundant number of large targets in highly damaged samples (Debode et al., 2007).
35In order to avoid the possible matrix effect, the influence of the target length on the signal was also studied with plasmids. In table 4, the increase of Cq value as a function of amplicon size in the double-dye probe format is observed. In SYBR® Green format, the influence was only visible with the 362 bp fragment.
4. Discussion
36Based on theoretical considerations linked to the fact that larger fragments should integrate a greater number of SYBR® Green molecules, one would expect a brighter signal when the target size increases. Theoretically, the results obtained in SYBR® Green should be opposite to those obtained with double-dye probes: the longer the amplicon, the more SYBR® Green molecules can intercalate between the two DNA strands. This differs from the double-dye system, where a single probe can hybridize between the two primers and where a single fluorophore emitting fluorescence is released, whatever the target size.
37When targets with an increasing size of fragments were amplified in real-time PCR, it was noted that the impact of the size increase on the Cq value was less pronounced in SYBR® Green format than with double-dye probes (Tables 3 and 4). However, SYBR® Green amplification of larger fragments did not lead to earlier detection as expected in theory. The target size effect was less visible than with double-dye probes because incorporation of a larger amount of SYBR® Green molecule in longer targets partially compensates for the lower levels of amplicons produced.
38Results obtained on non-damaged DNA (a template containing 100,000 copies of plasmid pl-RR) are presented in table 4: they show a similar upward trend in Cq value when the target size increases, which can no longer be due to a matrix effect.
39The possible influence of a non-optimal elongation step of the thermal program for the larger amplicons was checked to explain the observed phenomenon. The synthesis rate of the Taq polymerase is 60-150 nucleotides of elongation per second at 70-75 °C (Burrell, 1993). The velocity of the enzyme could be influenced by the concentrations of substrate and enzyme as well as temperature, based on the Michaelis-Menten principles (Schnell & Mendoza, 1997). Real-time PCR generally uses an extension temperature of 60 °C (or at least lower than 72 °C). This is due to the fact that, with TaqMan® probes, the probe has to remain bound during elongation in order to be degraded by the exonuclease activity of the Taq and to release the reporter dye, whose fluorescence will be measured by the real-time PCR thermocycler. It was also shown that it was possible to synthesize large fragments at 60 °C (Su et al., 1996) without adding time to the extension step.
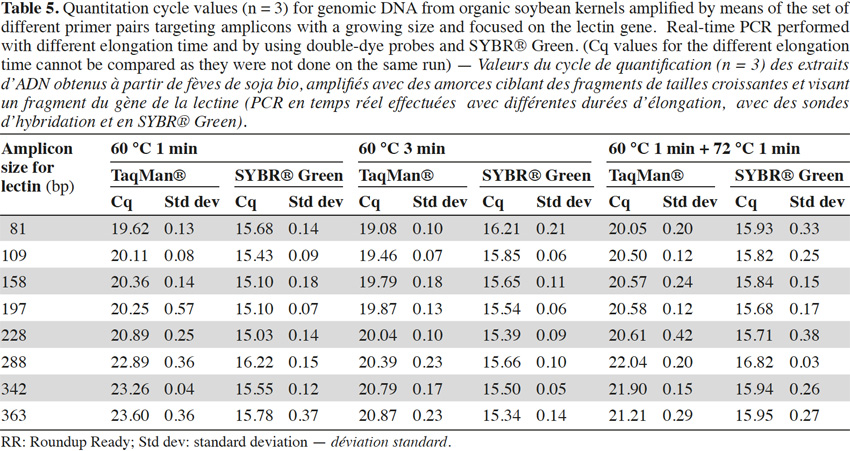
40To check the influence of the elongation step in our case, two modifications were made to the initial real-time PCR program used for the experiments (1 min at 60 °C for the hybridization and elongation steps). The first consisted of an increase of the hybridization/elongation step to 3 min at 60 °C, while the second consisted of an addition of 1 min at the optimum temperature of 72 °C after the minute at 60 °C. Tests were realized on genomic (Table 5) with the lectin target and showed a similar trend as described above, but the effect was less marked with a longer elongation step. This suggests that the loss of efficiency in real-time PCR with larger targets might be due to the fact that elongation at 60 °C is less rapid and will not always result in completed products that can be used as templates in further amplifications.
41Use of a lowered elongation temperature of for instance 60 °C instead of 72 °C in real-time PCR is crucial when the double-dye format is used, because the probe should still remain hybridized to its target when elongation starts. Therefore the results shown here indicate that when designing a real-time PCR in the double-dye format it is highly recommended to use targets which are as short as possible, especially if the elongation temperature lies far from the optimum temperature of the Taq polymerase used. Otherwise, an alternative is to set elongation time to a sufficiently long period to allow all templates targeted by the PCR test to be copied completely. This effect is less pronounced with SYBR® Green, but here too, especially if damaged DNA is used, it is better to use small targets.
42Acknowledgements
43The initial part of this research (Convention S-6062) was financed by the Belgian Federal Ministry of Public Health and “Fonds des matières premières”.
Bibliographie
Applied Biosystems, 2005. Real-Time PCR systems. Applied Biosystems 7900HT Fast Real-Time PCR System and 7300/7500 Real-Time PCR Systems. Chemistry Guide, http://www3.appliedbiosystems.com/cms/groups/mcb_marketing/documents/generaldocuments/cms_041440.pdf, (13/06/2012).
Bio-Rad Laboratories, 2006. Real-Time PCR Applications Guide, http://www.gene-quantification.de/real-time-pcr-guide-bio-rad.pdf, (13/06/2012).
Bruderer S. & Leitner K.E., 2003. Genetically Modified (GM) crops: molecular and regulatory details, http://www.bats.ch/gmo-watch/GVO-report140703.pdf, (20/06/2016).
Burrell M.,1993. Enzymes of molecular biology collection methods in molecular biology. Clifton, NJ, USA: Humana Press Inc.
Debode F., Janssen E. & Berben G., 2007. Physical degradation of genomic DNA of soybean flours does not impair relative quantification of its transgenic content. Eur. Food Res. Technol., 226, 273-280.
Farriol M. & Orta X., 2005. Influence of reagent formulation on mRNA quantification by RT-PCR using imported external standard curves. Acta Biochim. Pol., 52, 845-848.
Gryson N., 2010. Effect of food processing on plant DNA degradation and PCR-based GMO analysis: a review. Anal. Bioanal. Chem., 396, 2003-2022.
Holland P.M., Abramson D., Watson R. & Gelfand DH.,1991. Detection of specific polymerase chain reaction product by utilizing the 5'----3' exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. U.S.A., 88, 7276-7280.
Higuchi R., Fockler C., Dollinger G. & Watson R., 1993. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology, 11, 1026-1030.
Invitrogen. Product information, 2006. SYBR Green I Nucleic Acid Gel Stain. Cat. No. S7567, https://tools.thermofisher.com/content/sfs/manuals/mp07567.pdf, (08/02/17).
Kontanis E.-J. & Reed F.A., 2006. Evaluation of real-time PCR amplification efficiencies to detect PCR inhibitors. J. Forensic, 51, 795-804.
Meyer R., Chardonnens F., Hübner P. & Lüthy J., 1996. Polymerase chain reaction (PCR) in the quality and safety assurance of food: detection of soya in processed meat products. Z. Lebensmittel-Untersuchung Forschung, 203, 339-344.
Miller K., Smith R. & Storts D., 1996. Improved PCR amplification using TaqBeadTM hot start polymerase. Promega Notes Mag., 60, 2.
Opel K.L., Chung D. & McCord B.R., 2010. A study of PCR inhibition mechanisms using real time PCR. J. Forensic Sci., 55, 25-33.
Qiagen, 2011. QuantiFast® probe PCR handbook, http://www.qiagen.com/literature/handbooks/int/pcrlit.aspx#QuantiFast1, (13/06/2012).
Ririe K.M., Rasmussen R.P. & Wittwer C.T., 1997. Product differenciation by analysis of DNA melting curves during the polymerase chain reaction. Anal. Biochem., 245, 154-160.
Sambrook J., Fritsch E.F. & Maniatis T., 1989. Molecular cloning. A laboratory manual. 2nd ed. New York, USA: Cold Spring Harbor Laboratory Press.
Schnell S. & Mendoza C., 1997. Enzymological considerations for the theoretical description of the quantitative competitive polymerase chain reaction (QC-PCR). J. Theor. Biol., 184, 433-440.
Su X.-Z., Wu Y., Sifri C.D. & Wellems T.E., 1996. Reduced extension temperatures required for PCR amplification of extremely A+T-rich DNA. Nucleic Acids Res., 24, 1574-1575.
Windels P. et al., 2001. Characterisation of the Roundup Ready soybean insert. Eur. Food Res. Technol., 213, 107-112.