


- Startpagina tijdschrift
- Volume 15 (2011)
- numéro 1
- Revue bibliographique sur les méthodes d'analyse des polysaccharides structuraux des biomasses lignocellulosiques


Weergave(s): 16913 (200 ULiège)
Download(s): 1728 (15 ULiège)

Revue bibliographique sur les méthodes d'analyse des polysaccharides structuraux des biomasses lignocellulosiques

Nota's van de redactie
reçu le 21 mars 2009, accepté le 7 juin 2010
Résumé
Les polysaccharides structuraux et la lignine représentent l'essentiel des biomasses lignocellulosiques. La composition de ces polymères, notamment la cellulose, les hémicelluloses et les pectines, peut être effectuée par diverses méthodes globales et/ou sélectives de ces polymères. Les méthodes globales étudiées dans cette synthèse sont celles de « Van Soest », « Prosky », « Uppsala », « Selvendran » et des « polysaccharides non amylacés ». Afin de distinguer les différences, les avantages et les inconvénients de ces méthodes globales, celles-ci sont décrites et comparées. La méthode de Van Soest est celle présentant le meilleur compromis entre les données obtenues sur la nature des polysaccharides structuraux constituant les biomasses lignocellulosiques et le temps d'analyse pour parvenir à obtenir cette information. En adaptant cette méthode, elle pourrait apporter des données sur la quantité de pectines (exprimées en acide galacturonique), sur la composition en monosaccharides des polysaccharides structuraux et sur la quantité d'hydrates de carbone non structuraux des biomasses lignocellulosiques. Quelques méthodes de dosage sélectives d'un ou deux polysaccharides structuraux comme la cellulose, les xyloglucanes, les xylanes, les β-glucanes, les mannanes et les pectines sont également présentées dans cette synthèse.
Abstract
Review on analytical methods for lignocellulosic biomass structural polysaccharides. Structural polysaccharides and lignin represent the major part of the lignocellulosic biomass. The characterization of these structural polymers, like cellulose, hemicelluloses and pectins, can be carried out by various global and/or polymer-selective methods. The global methods studied in this review are those of “Van Soest”, “Prosky”, “Uppsala”, “Selvendran” and “non-starch polysaccharides”. These global methods are described and compared in order to enlighten their differences, advantages and disadvantages. The Van Soest method is the one presenting the best compromise between the data obtained on the nature of structural polysaccharides constituting the lignocellulosic biomass and the time of analysis required to obtain this piece of information. By adapting this method, it could also give information on the quantity of pectins (expressed as galacturonic acid), on the monosaccharidic composition of the structural polysaccharides and on the quantity of non structural carbohydrates of the lignocellulosic biomass. Some methods selective to one or two structural polysaccharides like cellulose, xyloglucans, xylans, β-glucans, mannanes and pectins are also presented in this review.
Inhoudstafel
1. Introduction
1Les hydrates de carbone des biomasses lignocellulosiques sont séparés en deux catégories : les hydrates de carbone non structuraux comme les monosaccharides, les oligosaccharides et les polysaccharides de réserves, et les hydrates de carbone structuraux (dont la dénomination plus commune de « polysaccharides structuraux » est utilisée par la suite dans cet article) (Southgate, 1995 ; Carpita et al., 2000). La paroi cellulaire des biomasses lignocellulosiques est constituée d'une grande diversité de ces polysaccharides structuraux qui sont divisés en différentes catégories : la cellulose, les hémicelluloses (xyloglucanes, xylanes, β-glucanes et mannanes) et les pectines (homogalacturonanes, xylogalacturonanes, rhamnogalacturonanes type I et type II) (Carpita et al., 2000). Ces composés sont liés à des protéines pariétales (riches en acides aminés, soit de type hydroxyproline, soit de type thréonine-hydroxyproline, soit de type glycine) et à des polyphénols, plus particulièrement la lignine qui est un polymère constitué d'unités phénylpropanes telles que les alcools de type p-hydroxyphényle (dont l'alcool p-coumarylique), syringyle (dont l'alcool synapilique) et guaïacyle (dont l'alcool coniférylique) (Carpita et al., 2000 ; Grabber, 2005). Les polysaccharides structuraux et la lignine sont les constituants les plus abondants dans la paroi cellulaire (Ebringerová et al., 2005). Tous les constituants des parois végétales des biomasses lignocellulosiques (Figure 1) sont qualifiés de fibres et plus spécifiquement de fibres alimentaires dans le domaine de la nutrition (FAO, 2007 ; Ampuero, 2008). La FAO définit les fibres alimentaires comme « des polymères glucidiques avec un degré de polymérisation (DP) non inférieur à trois, qui ne sont ni digérés ni absorbés dans l'intestin grêle. Un degré de polymérisation non inférieur à trois est destiné à exclure les mono- et disaccharides et non à refléter le DP moyen du mélange. Si elles sont d'origine végétale, les fibres alimentaires peuvent comprendre des fractions de lignine et/ou d'autres composants s'ils sont associés avec des polysaccharides dans les parois cellulaires végétales et si ces composants sont quantifiés par la méthode d'analyse gravimétrique qui a été adoptée pour l'analyse des fibres alimentaires (AOAC) » (FAO, 2007).
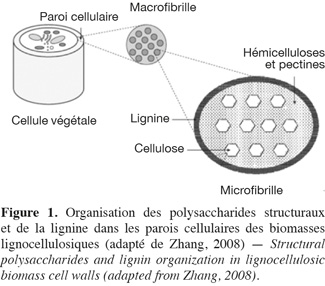
2Deux catégories de méthodes existent afin de caractériser la nature des polysaccharides structuraux des biomasses lignocellulosiques : les méthodes globales et les méthodes sélectives.
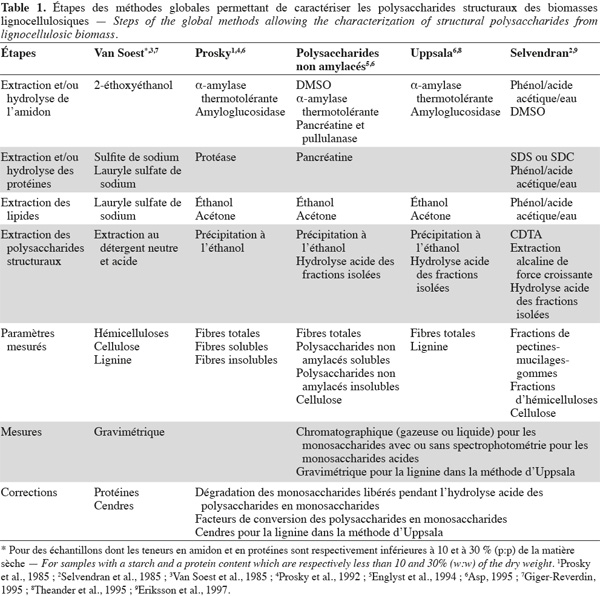
3Les méthodes globales permettent de caractériser des ensembles de polysaccharides structuraux sans en distinguer la nature dans chaque ensemble isolé. Ces polysaccharides sont séparés soit sous forme de fibres alimentaires totales, soit divisés en fibres alimentaires solubles et insolubles dans l'eau. Il faut noter que la lignine, les protéines pariétales, les tannins et d'autres composants liés aux polysaccharides structuraux se trouvent également dans les fibres alimentaires car ils font partie intégrante de ces fractions de fibres alimentaires (Dorleans et al., 1996 ; Carpita et al., 2000 ; FAO, 2007). Les principales méthodes globales sont la méthode aux détergents neutre et acide de Van Soest et al. (1985), la méthode des fibres alimentaires de Prosky et al. (1992), la méthode des polysaccharides non amylacés d'Englyst et al. (1994), la méthode des fibres alimentaires d'Uppsala (Theander et al., 1995) et la méthode alcaline de Selvendran et al. (1985).
4Les méthodes sélectives isolent sélectivement un ou deux polysaccharides structuraux, comme la cellulose (Sun et al., 1995), les xylanes (Izydorczyk et al., 1998a ; Izydorczyk et al., 1998b ; Genestie, 2006), les xyloglucanes (Cutillas-Iturralde et al., 1998), les β-glucanes (Izydorczyk et al., 1998a ; Izydorczyk et al., 1998b), les mannanes (Selvendran et al., 1985) et les pectines (Selvendran et al., 1985 ; Le Goff et al., 2001).
5L'objectif de cette synthèse est de déterminer la méthode présentant le meilleur compromis entre les données obtenues sur la nature (la structure et la composition en monosaccharides) des polysaccharides structuraux constituant la biomasse lignocellulosique et le temps d'analyse pour parvenir à obtenir cette information. Les hydrates de carbone non structuraux représentent une des interférences majeures lors de l'analyse des polysaccharides structuraux des biomasses lignocellulosiques. La purification des polysaccharides structuraux par une méthode suffisamment sélective est donc nécessaire pour déterminer la nature de ces polysaccharides des biomasses lignocellulosiques.
6Les méthodes globales de Van Soest, de Prosky, des polysaccharides non amylacés, d'Uppsala et de Selvendran seront présentées et comparées dans cette revue. Les méthodes sélectives à la cellulose, aux xylanes, aux xyloglucanes, aux β-glucanes, aux mannanes et aux pectines des biomasses lignocellulosiques seront également présentées dans cette revue.
2. Description des méthodes d'analyse des polysaccharides structuraux des biomasses lignocellulosiques
2.1. Méthode aux détergents neutre et acide de Van Soest
7La méthode aux détergents neutre et acide de Van Soest et al. (1985) (Figure 2, Tableaux 1 et 2) fractionne les fibres de la biomasse lignocellulosique par des extractions successives par voie chimique et quantifie par gravimétrie les fibres insolubles sèches récupérées après filtration.
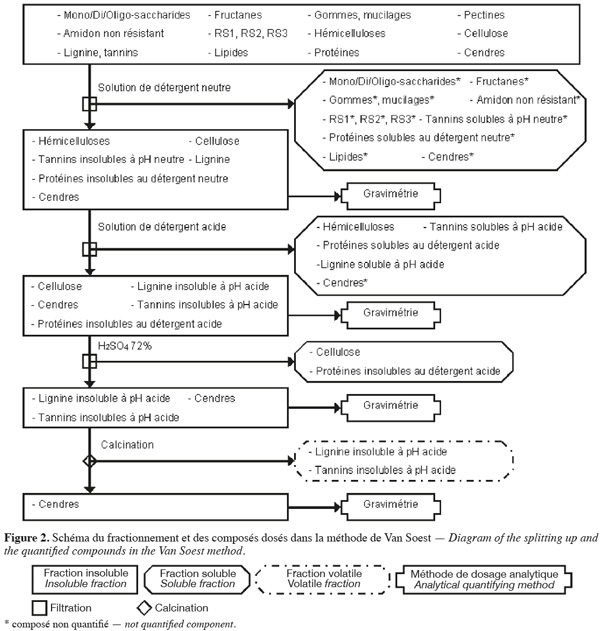

8La méthode de Van Soest (Van Soest et al., 1985 ; Van Soest et al., 1991 ; Mertens, 2002 ; Ampuero, 2008) permet d'extraire les hydrates de carbone non structuraux, les pectines, les gommes, les mucilages1, les tannins solubles à pH neutre, les lipides, les protéines solubles au détergent neutre et certaines cendres à l'aide d'une solution de détergent neutre, en excès, agissant sur l'échantillon pendant 1 h à 100 °C avec un pH de 7 ± 0,05. Cette solution de détergent neutre est composée de Na2HPO4, de tétraborate de sodium, de 2-éthoxyéthanol (qui extrait l'amidon), d'EDTA de sodium (qui extrait les pectines), de lauryle sulfate de sodium (qui extrait les protéines et les lipides) et de sulfite de sodium (qui hydrolyse les protéines). La fraction extraite est séparée des fibres insolubles au détergent neutre (NDF) par filtration. La fraction NDF est constituée de cellulose, d'hémicelluloses, de lignine, de tannins insolubles à pH neutre, de protéines insolubles au détergent neutre et de certaines cendres.
9Parallèlement (ou séquentiellement) à l'action du détergent neutre, une solution de détergent acide, en excès, permet d'extraire les hémicelluloses, les tannins solubles en milieu acide (présents en très faible quantité), la lignine soluble en milieu acide (présente en très faible quantité), les protéines solubles au détergent acide et certaines cendres. Cette solution de détergent acide est composée de bromure de triméthyl-cétyl-ammonium et de H2SO4 1 N et agit pendant 1 h à 100 °C. La fraction extraite est séparée des fibres insolubles au détergent acide (ADF) par filtration. La fraction ADF est constituée de cellulose, de lignine insoluble en milieu acide, de tannins insolubles en milieu acide, des protéines insolubles au détergent acide et de certaines cendres. La différence entre les NDF et les ADF (en corrigeant par la teneur en protéines et en cendres) correspond à la quantité d'hémicelluloses, de lignine soluble en milieu acide et de tannins solubles en milieu acide.
10La cellulose et les protéines insolubles au détergent acide sont extraites en traitant la fraction ADF au H2SO4 72 % pendant 3 h à 20-23 °C. La fraction extraite est séparée du résidu insoluble par filtration. La quantité de cellulose correspond à la perte de masse insoluble de la fraction ADF (en corrigeant par la teneur en protéines) durant cette hydrolyse. Le résidu insoluble est composé de lignine insoluble en milieu acide, de tannins insolubles en milieu acide et de cendres.
11La lignine insoluble en milieu acide et les tannins insolubles en milieu acide (présents en très faible quantité) sont volatilisés par calcination. Le résidu insoluble obtenu après calcination est composé de cendres. La lignine insoluble en milieu acide avec les tannins insolubles en milieu acide sont quantifiés par la perte de masse durant la calcination.
12Afin de limiter l'incertitude sur la quantification des résidus insolubles, les valeurs gravimétriques obtenues pour tous les résidus insolubles doivent être corrigées par la teneur en protéines et en cendres. Les protéines sont quantifiées par un micro-Kjeldahl ou une analyse élémentaire sur base de l'azote en appliquant un facteur de conversion N (gN) x 6,25 (gprotéines.gN-1) (AOAC, 1990). Les cendres sont dosées par calcination à 525 °C (Van Soest et al., 1985). Les quantités de cellulose et d'hémicelluloses peuvent être respectivement surestimées et sous-estimées dans le cas d'une contamination des ADF par des hémicelluloses. Cette contamination peut atteindre la valeur de 15,4 % des polysaccharides structuraux des ADF (Morrison, 1980 ; Jung, 1997). Les quantités de cellulose et d'hémicelluloses des biomasses lignocellulosiques possédant une teneur importante en protéines sont surestimées si la valeur respectivement des NDF et des ADF n'est pas corrigée par leurs teneurs en protéines (Jung, 1997). La quantité d'hémicelluloses des biomasses lignocellulosiques possédant une teneur importante en lignine soluble à pH acide est surestimée car cette lignine soluble est contenue dans les NDF, mais elle n'est pas contenue dans les ADF (Jung, 1997). Les ADF n'ayant pas subi de traitement au détergent neutre contiennent certaines pectines qui ne sont pas contenues dans les ADF ayant subi un traitement préalable au détergent neutre. Les hémicelluloses des ADF n'ayant pas subi de traitement au détergent neutre sont sous-estimées si la biomasse lignocellulosique possède une teneur importante en pectines (Ampuero, 2008).
13La méthode de Van Soest ne s'applique qu'à des échantillons dont les teneurs en amidon, en protéines et en lipides sont respectivement inférieures à 10, à 30 et à 10 % (p:p) de la matière sèche. Les NDF sont surestimées lorsque l'échantillon contient trop d'amidon et de protéines. S'il contient trop de lipides, le détergent neutre ou acide s'associe préférentiellement à la phase lipidique et perd de son efficacité, donnant des valeurs biaisées pour les NDF et les ADF (Van Soest et al., 1985 ; Van Soest et al., 1991 ; Giger-Reverdin, 1995). Dans le cas d'un excès de lipides, il suffit d'extraire préalablement les lipides de l'échantillon pour pouvoir appliquer la méthode de Van Soest. Dans le cas d'un excès d'amidon et de protéines, des α-amylases et des protéases sont respectivement utilisées (Giger-Reverdin, 1995). Différentes autres variantes de la méthode aux détergents neutre et acide de Van Soest ont été développées : ajout d'α-amylases thermotolérantes au détergent neutre pour hydrolyser l'amidon (Mertens, 2002), ajout de protéases préalablement à l'attaque au détergent neutre pour hydrolyser les protéines (Dorleans et al., 1996), utilisation de quantités plus faibles de détergent neutre pour utiliser un moindre excès de détergent neutre (Chai et al., 1998).
2.2. Méthode des fibres alimentaires de Prosky
14La méthode des fibres alimentaires de Prosky (Prosky et al., 1985 ; Prosky et al., 1992) (Figure 3, Tableaux 1 et 2) est basée sur des hydrolyses enzymatiques successives de la biomasse lignocellulosique afin d'extraire les molécules qui ne font pas partie des fibres alimentaires. Les fibres alimentaires sont quantifiées par gravimétrie.
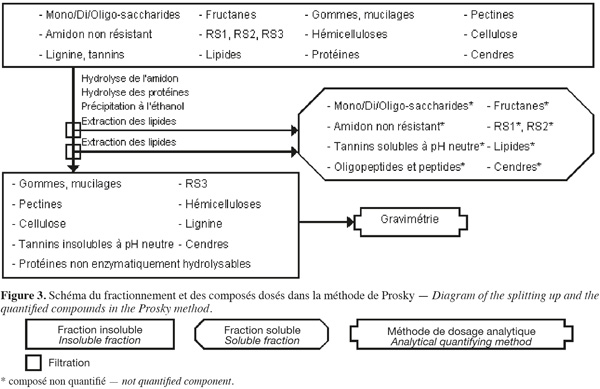
15La méthode de Prosky (Prosky et al., 1985 ; Prosky et al., 1992 ; Asp, 1995 ; Englyst et al., 1996 ; McCleary et al., 2002 ; FAO, 2007) commence par une extraction des lipides, si l'échantillon en contient plus de 10 % (p:p). Cette méthode permet d'extraire ou d'hydrolyser les mono/di/oligo-saccharides, les fructanes, les gommes, les mucilages, l'amidon non résistant (accessible et non résistant à l'hydrolyse par l'α-amylase pancréatique), le RS1 (amidon inaccessible physiquement à l'α-amylase pancréatique), le RS2 (amidon non gélatinisé et fortement résistant à l'hydrolyse par l'α-amylase pancréatique), les tannins solubles à pH neutre, les protéines enzymatiquement hydrolysables et certaines cendres à l'aide de, successivement, une α-amylase thermotolérante à 100 °C pendant 30 min, une protéase à 60 °C pendant 30 min et une amyloglycosidase à 60 °C pendant 60 min. Les fibres alimentaires totales sont isolées par une précipitation à l'éthanol aqueux 78 % (v:v) suivie d'une filtration sur diatomite. Les fibres alimentaires totales insolubles sont ensuite rincées deux fois à l'éthanol aqueux 95 % (v:v) et deux fois à l'acétone. Les étapes de précipitation à l'éthanol, de rinçage à l'éthanol et à l'acétone extraient les lipides. Les fibres alimentaires totales sont composées de cellulose, d'hémicelluloses, du RS3 (amidon gélatinisé et fortement résistant à l'hydrolyse par l'α-amylase), des pectines (sauf celles qui sont très hydrosolubles), des gommes (sauf celles qui sont très hydrosolubles), des mucilages (sauf ceux qui sont très hydrosolubles), de la lignine, des tannins insolubles à pH neutre, des protéines non enzymatiquement hydrolysables et de cendres.
16Afin de limiter l'incertitude sur le résultat des fibres alimentaires totales, chaque analyse est effectuée en double. Sur l'une des répétitions, l'azote est dosé par un micro-Kjeldahl ou une analyse élémentaire pour corriger le contenu en protéines dans la fraction insoluble, en appliquant un facteur de conversion N (gN) x 6,25 (gprotéines.gN-1) (AOAC, 1990). Sur l'autre répétition, les cendres sont quantifiées par calcination à 525 °C dans la fraction insoluble. Le même protocole est appliqué en parallèle sur deux témoins blancs (exempt d'échantillon) pour déterminer les contributions gravimétriques des différentes solutions utilisées (Prosky et al., 1985 ; Prosky et al., 1992).
17Dans une autre variante de la méthode de Prosky, les fibres alimentaires totales sont séparées en fibres alimentaires insolubles et solubles dans l'eau à pH neutre. Au lieu de réaliser l'étape de précipitation à l'éthanol aqueux 78 % (v:v), la solution est directement filtrée sur diatomite afin de récupérer les fibres alimentaires solubles dans le filtrat et les fibres alimentaires insolubles dans le résidu insoluble (Prosky et al., 1992). Dans une autre variante de la méthode de Prosky, le temps d'analyse est réduit en diminuant le volume total de filtration par l'utilisation de volumes de réactifs plus faibles et en n'ajustant pas le pH lors de l'utilisation de la protéase (Lee et al., 1992).
2.3. Méthode de dosage des polysaccharides non amylacés (NSP) d'Englyst
18La méthode de dosage des polysaccharides non amylacés d'Englyst et al. (1994) (Figure 4, Tableaux 1 et 2) extrait séquentiellement par voie enzymatique et chimique différentes molécules de la biomasse lignocellulosique qui ne font pas partie des polysaccharides non amylacés. Les polysaccharides non amylacés sont quantifiés par chromatographie et avec ou sans spectrophotométrie après hydrolyse acide.
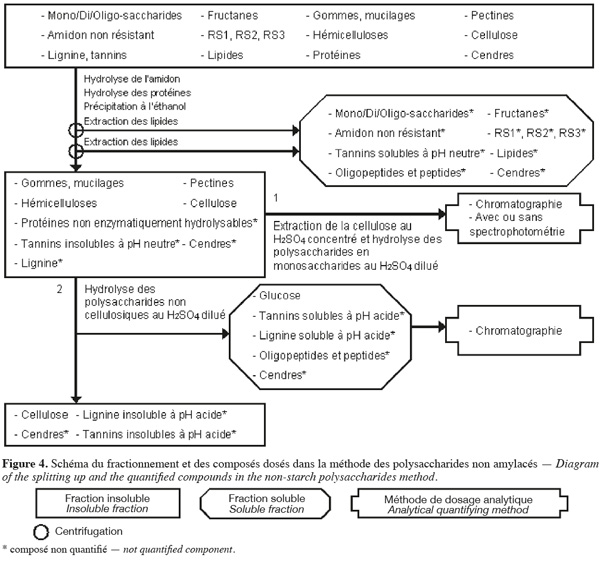
19La méthode des polysaccharides non amylacés (Englyst et al., 1994 ; Asp, 1995 ; Englyst et al., 1996) commence par une extraction des lipides, si l'échantillon en contient plus de 10 % (p:p). Cette méthode permet d'extraire ou d'hydrolyser les mono/di/oligo-saccharides, les fructanes, l'amidon non résistant, le RS1, le RS2, le RS3, les tannins solubles à pH neutre, les protéines enzymatiquement hydrolysables et certaines cendres à l'aide, successivement, du diméthylsulfoxide (DMSO) pendant 30 min à 100 °C, une α-amylase thermotolérante à 100 °C pendant 10 min et un mélange de pancréatine et de pullulanase pendant 30 min à 50 °C. Les polysaccharides non amylacés totaux sont isolés par une précipitation à l'éthanol aqueux 80 % (v:v) à pH 2 suivie d'une centrifugation. Les polysaccharides non amylacés totaux insolubles sont ensuite rincés une fois à l'éthanol aqueux 85 % (v:v) acidifié, une fois à l'éthanol aqueux 99,5 % (v:v) et une fois à l'acétone. Les étapes de précipitation à l'éthanol, de rinçage à l'éthanol et à l'acétone extraient les lipides. Les polysaccharides non amylacés totaux sont composés de cellulose, d'hémicelluloses, des pectines (sauf celles qui sont très hydrosolubles), des gommes (sauf celles qui sont très hydrosolubles), des mucilages (sauf ceux qui sont très hydrosolubles), de la lignine, des tannins insolubles à pH neutre, des protéines non enzymatiquement hydrolysables et de cendres.
20Les polysaccharides non amylacés obtenus sont hydrolysés en monosaccharides à l'aide d'H2SO4 12 M pendant 30 min à 35 °C. De l'eau est ensuite ajoutée afin de diluer le H2SO4 jusqu'à 2 M et de poursuivre l'hydrolyse des polysaccharides non amylacés en monosaccharides pendant 1 h à 100 °C. Les monosaccharides sont quantifiés soit par HPLC, soit par GC pour les monosaccharides neutres en les dérivatisant en acétate d'alditol et par spectrophotométrie pour les monosaccharides acides (acides uroniques) en les transformant en acide 5-formyl-2-furoïque. Cet acide est dosé après réaction avec du 3,5-diméthylphénol en faisant la différence entre l'absorbance à 450 nm et à 400 nm. La somme de tous les monosaccharides déterminés après hydrolyse correspond aux polysaccharides non amylacés (Englyst et al., 1994).
21La cellulose peut aussi être quantifiée en effectuant le même protocole en double. Afin d'hydrolyser les polysaccharides non cellulosiques, pour l'une des deux analyses, le culot correspondant aux polysaccharides non amylacés totaux est soumis à une étape d'hydrolyse au H2SO4 2 M pendant 60 min à 100 °C. Le glucose libéré lors de cette étape est analysé par chromatographie gazeuse ou liquide. La quantité de cellulose correspond à la différence du glucose entre l'échantillon « normal » (glucose venant des polysaccharides non amylacés totaux) et l'échantillon ayant subi le traitement alternatif (glucose venant des polysaccharides non cellulosiques) (Englyst et al., 1994).
22La dégradation des monosaccharides libérés pendant l'hydrolyse acide des polysaccharides en monosaccharides analysés par GC, la dégradation des monosaccharides libérés pendant l'hydrolyse acide des polysaccharides en monosaccharides analysés par HPLC et le rendement de dérivatisation des monosaccharides neutres analysés par GC sont déterminés en utilisant comme référence un mélange de standards de monosaccharides dans du H2SO4 2 M. Ce mélange de standards est traité en parallèle avec les échantillons issus de l'étape de dilution du H2SO4 concentré. Un standard interne est ajouté après l'hydrolyse au H2SO4 dilué (Englyst et al., 1994). La quantification des polysaccharides non amylacés prend également en compte le facteur de conversion des polysaccharides en monosaccharides (gain d'une molécule d'eau par l'hydrolyse), à savoir 89 % pour les monosaccharides neutres et 91 % pour les monosaccharides acides (Englyst et al., 1994).
23Dans une variante de la méthode de dosage des polysaccharides non amylacés, les polysaccharides non amylacés totaux sont séparés en polysaccharides non amylacés insolubles et solubles dans l'eau à pH neutre. Au lieu de réaliser l'étape de précipitation à l'éthanol aqueux 80 % (v:v) à pH 2, la solution est directement centrifugée afin de récupérer les polysaccharides non amylacés solubles dans le filtrat et les polysaccharides non amylacés insolubles dans le résidu insoluble (Englyst et al., 1994).
2.4. Méthode des fibres alimentaires d'Uppsala
24La méthode des fibres alimentaires d'Uppsala (Theander et al., 1995) (Figure 5, Tableaux 1 et 2) extrait séquentiellement par voie enzymatique et chimique différentes molécules de la biomasse lignocellulosique qui ne font pas partie des fibres alimentaires. Les fibres alimentaires sont quantifiées par chromatographie, spectrophotométrie et gravimétrie après hydrolyse acide.
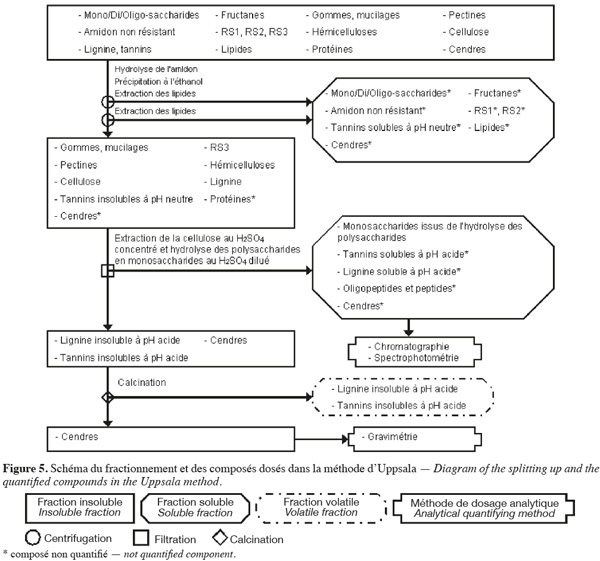
25La méthode des fibres alimentaires d'Uppsala (Theander et al., 1995 ; Asp, 1995 ; Englyst et al., 1996 ; FAO, 2007) commence par une extraction des lipides, si l'échantillon en contient plus de 5 % (p:p). Cette méthode permet d'extraire ou d'hydrolyser les mono/di/oligo-saccharides, les fructanes, l'amidon non résistant, le RS1, le RS2, les tannins solubles à pH neutre et certaines cendres à l'aide de, successivement, une α-amylase thermotolérante à 100 °C pendant 60 min et une amyloglycosidase à 60 °C pendant 6 h. Les fibres alimentaires totales sont isolées par une précipitation à l'éthanol aqueux 80 % (v:v) suivie d'une centrifugation. Les fibres alimentaires totales insolubles sont ensuite rincées une fois à l'éthanol aqueux 99,5 % (v:v) et deux fois à l'acétone. Les étapes de précipitation à l'éthanol, de rinçage à l'éthanol et à l'acétone extraient les lipides. Les fibres alimentaires totales sont composées de cellulose, d'hémicelluloses, du RS3, des pectines (sauf celles qui sont très hydrosolubles), des gommes (sauf celles qui sont très hydrosolubles), des mucilages (sauf ceux qui sont très hydrosolubles), de la lignine, des tannins insolubles à pH neutre, des protéines et des cendres.
26Les fibres alimentaires obtenues sont hydrolysées en monosaccharides à l'aide d'H2SO4 12 M pendant 60 min à 30 °C. De l'eau est ensuite ajoutée afin de diluer le H2SO4 jusqu'à 0,47 M et de poursuivre l'hydrolyse des fibres alimentaires en monosaccharides pendant 1 h à 125 °C. La solution est filtrée afin de récupérer les monosaccharides dans le filtrat. Les monosaccharides du filtrat sont quantifiés par GC en les dérivatisant en acétate d'alditol, alors que les monosaccharides uroniques (acides uroniques) sont quantifiés par spectrophotométrie selon le même protocole que dans la méthode des polysaccharides non amylacés. Le résidu insoluble de filtration permet de quantifier par gravimétrie la lignine insoluble à pH acide avec les tannins insolubles à pH acide (présents en très faible quantité), qui forment ensemble la « lignine de Klason ». La lignine insoluble à pH acide et les tannins insolubles à pH acide sont volatilisés par calcination. Le résidu insoluble obtenu après calcination est composé de cendres. La lignine insoluble en milieu acide et les tannins insolubles en milieu acide sont quantifiés par la perte de masse durant la calcination à 500 °C. La somme de tous les monosaccharides déterminés après hydrolyse et de la lignine de Klason correspond aux fibres alimentaires (Theander et al., 1995).
27La dégradation des monosaccharides libérés pendant l'hydrolyse acide des polysaccharides en monosaccharides analysés par GC et le rendement de dérivatisation des monosaccharides neutres analysés par GC sont déterminés en utilisant comme référence un mélange de standards des monosaccharides dans du H2SO4 0,47 M. Ce mélange de standards est traité en parallèle avec les échantillons issus de l'étape de dilution du H2SO4 concentré. Un standard interne est ajouté à l'étape d'hydrolyse au H2SO4 dilué. La quantification des polysaccharides non amylacés prend également en compte le facteur de conversion des polysaccharides en monosaccharides (gain d'une molécule d'eau par l'hydrolyse), à savoir 91 % pour les monosaccharides acides, 88 % pour les pentoses et les déoxyhexoses et 90 % pour les autres hexoses (Theander et al., 1995). Afin de limiter l'incertitude sur la quantification de la lignine de Klason, la valeur gravimétrique du résidu insoluble obtenu après l'hydrolyse acide doit être corrigée par la teneur en protéines résiduelles contenues dans ce résidu. Les protéines sont quantifiées par un micro-Kjeldahl ou une analyse élémentaire sur base de l'azote en appliquant un facteur de conversion N (gN) x 6,25 (gprotéines.gN) (Hatfield et al., 1994).
2.5. Méthode alcaline de Selvendran
28La méthode alcaline de Selvendran et al. (1985) (Figure 6, Tableaux 1 et 2) extrait séquentiellement par voie chimique différentes fractions spécifiques de la biomasse lignocellulosique. Les polysaccharides structuraux sont quantifiés par chromatographie et par spectrophotométrie après hydrolyse acide. Cette méthode se démarque de celles décrites ci-dessus du fait qu'elle permet un fractionnement sélectif des polysaccharides structuraux (plus de deux polysaccharides structuraux) en fonction de leur nature.
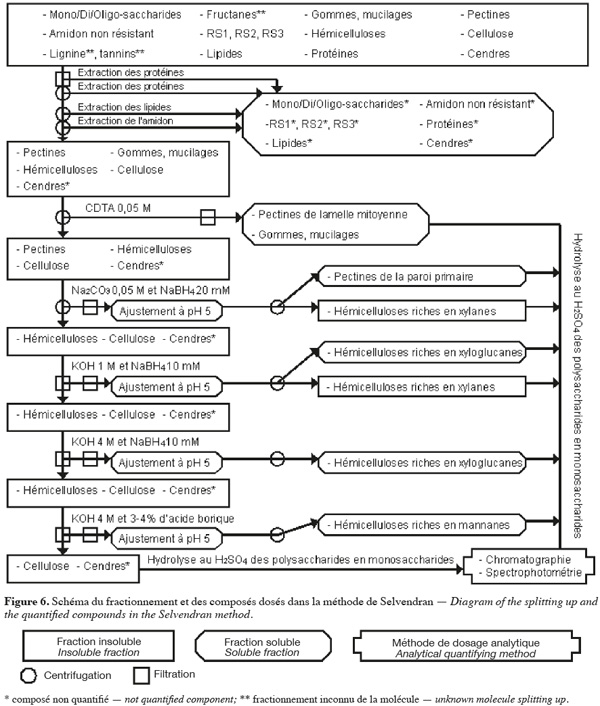
29Les protéines de l'échantillon sont d'abord éliminées par un traitement au déoxycholate de sodium (SDC) aqueux à 1 % (v:v) ou au dodécyl sulphate de sodium (SDS) aqueux à 1,5 %, contenant 5 mM de Na2S2O5 (pour limiter la formation de produits d'oxydation des polyphénols) pendant 5 min. La solution est filtrée et cette étape est répétée sur le résidu insoluble. Ce résidu insoluble est broyé à une taille inférieure à 3 mm dans du déoxycholate de sodium (SDC) aqueux à 0,5 % (v:v) ou du dodécyl sulphate de sodium (SDS) aqueux à 0,5 %, contenant 3 mM de Na2S2O5 pendant 5 min. La solution est centrifugée et cette étape est répétée sur le résidu insoluble. Les filtrats et les surnageants obtenus jusqu'à cette étape contiennent des protéines, des pectines très hydrosolubles, certaines cendres, des composés intracellulaires comme les mono/di/oligo-saccharides. Les protéines résiduelles, les lipides, les pigments et une partie de l'amidon sont extraits du résidu insoluble par deux traitements avec une solution phénol/acide acétique/eau (2:1:1 ; p/v/v). La solution est centrifugée et cette étape est répétée sur le résidu insoluble. L'amidon est extrait du résidu insoluble par du DMSO aqueux à 90 % (v/v) à 20 °C pendant 16 h. Le résidu insoluble est centrifugé et lavé six fois à l'eau afin d'obtenir les composants pariétaux purifiés (Selvendran et al., 1985).
30Les pectines et les hémicelluloses riches en xylanes sont extraites du résidu insoluble. Premièrement, ce dernier est traité au N,N,N',N'-tétraacétate cyclohexanediamine (CDTA) de sodium 0,05 M à 21 °C et à pH 6,5 pendant 6 h. Le CDTA de sodium est un agent chélatant qui extrait principalement les substances pectiques de la lamelle mitoyenne et qui, à une température de 20 °C, est aussi efficace que des agents chélatants conventionnels à haute température. La solution est centrifugée et cette étape est répétée sur le résidu insoluble, mais la durée à appliquer est de 2 h. Les deux surnageants contenant des pectines de la lamelle mitoyenne sont filtrés, dialysés et concentrés. Deuxièmement, le résidu insoluble est soumis au Na2CO3 0,05 M et au NaBH4 20 mM (pour protéger les polysaccharides de la β-élimination) pendant 16 h à 1 °C. La solution est centrifugée et extraite une deuxième fois au Na2CO3 0,05 M et au NaBH4 20 mM pendant 3 h à 1 °C. Le résidu insoluble correspond aux composants pariétaux purifiés et dépectinés. Les deux surnageants contenant des pectines de la paroi primaire et d'hémicelluloses riches en xylanes sont filtrés. Le pH de ces filtrats est ajusté à pH 5 par de l'acide acétique afin de précipiter les hémicelluloses riches en xylanes. Après centrifugation, les surnageants de pectines sont dialysés et concentrés (Selvendran et al., 1985 ; Southgate, 1995).
31Les hémicelluloses (xylanes résiduels, xyloglucanes, β-glucanes et mannanes) et la cellulose sont isolées du résidu insoluble des composants pariétaux purifiés et dépectinés. Dans un premier temps, le résidu insoluble est soumis à la KOH 1 M désoxygénée et au NaBH4 10 mM pendant 2 h à 1 °C. La solution est filtrée. Le résidu insoluble est extrait une deuxième fois à la KOH 1 M désoxygénée et au NaBH4 10 mM pendant 2 h à 21 °C sous azote ou argon et cette solution est filtrée. Le pH des deux filtrats contenant les hémicelluloses extraites est ajusté à pH 5 par de l'acide acétique afin de précipiter les hémicelluloses riches en xylanes. Ces solutions sont centrifugées afin d'obtenir les hémicelluloses riches en xylanes dans le culot et les hémicelluloses riches en xyloglucanes dans le surnageant. Le surnageant avec les hémicelluloses riches en xyloglucanes est dialysé et concentré. Dans un deuxième temps, le résidu insoluble obtenu après deux traitement à la KOH 1 M est soumis à la KOH 4 M désoxygénée et au NaBH4 10 mM pendant 2 h à 21 °C sous argon ou azote. La solution est filtrée et le résidu insoluble est soumis une deuxième fois à la KOH 4 M désoxygénée et au NaBH4 10 mM pendant 2 h à 21 °C sous azote ou argon avec de l'acide borique 3-4 %. Les deux filtrats contenant des hémicelluloses sont filtrés. Le pH des filtrats est ajusté à pH 5 par de l'acide acétique. Ces nouveaux filtrats sont dialysés et concentrés. Le premier filtrat est composé d'hémicelluloses riches en xyloglucanes, alors que le deuxième filtrat est constitué d'hémicelluloses riches en mannanes. Le résidu insoluble obtenu est essentiellement composé de cellulose, de lignine et de cendres (Selvendran et al., 1985 ; Southgate, 1995).
32Après les étapes de dépectination de l'échantillon, il peut être délignifié pour rendre le réseau de cellulose et d'hémicelluloses plus accessible à l'extraction alcaline (Genestie, 2006). La délignification a lieu à 70 °C par du NaClO2 et de l'acide minéral ou organique pour une durée entre 1 et 4 h selon le degré de lignification de l'échantillon. Cette étape de délignification a plusieurs effets sur l'échantillon analysé. Premièrement, elle n'est que partielle. Elle est de 60 % si cette étape dure 2 h. Deuxièmement, elle extrait de faibles quantités, aux environs 5 à 10 %, des polysaccharides de l'échantillon. Troisièmement, elle extrait partiellement les protéines pariétales (Selvendran et al., 1985).
33Les polysaccharides de toutes les fractions obtenues et neutralisées sont caractérisés et quantifiés en suivant le même protocole que dans la méthode d'Uppsala, c'est-à-dire à partir de l'hydrolyse acide au H2SO4 12 M (Selvendran et al., 1985 ; Eriksson et al., 1997).
2.6. Méthodes sélectives d'un ou de deux polysaccharides structuraux des biomasses lignocellulosiques
34De nombreuses méthodes d'analyse sélective d'un ou de deux polysaccharides structuraux des biomasses lignocellulosiques existent dans la littérature. Certaines de ces méthodes d'analyse sont décrites ci-dessous pour la cellulose (Sun et al., 1995), les xyloglucanes (Cutillas-Iturralde et al., 1998), les xylanes (Izydorczyk et al., 1998a ; Izydorczyk et al., 1998b ; Genestie, 2006), les β-glucanes (Izydorczyk et al., 1998a ; Izydorczyk et al., 1998b), les mannanes (Selvendran et al., 1985) et les pectines (Selvendran et al., 1985 ; Le Goff et al., 2001).
35Cellulose. La méthode de Sun et al. (1995) isole à partir de paille de froment la cellulose en plusieurs étapes d'extraction chimique. Deux des étapes intermédiaires permettent d'isoler une fraction riche en pectines et une fraction riche en hémicelluloses.
36Les composées intracellulaires, les hydrates de carbone non structuraux et une partie de lignine, d'hémicelluloses, des pectines et des protéines sont extraits par une étape de prétraitement de l'échantillon à la NaOH 1,5 % pendant 144 h à 20 °C. Les pectines résiduelles dans l'échantillon sont extraites par un agent chélatant, l'oxalate d'ammonium à 0,25 % pendant 4 h à 85 °C. La lignine résiduelle est éliminée par une extraction au NaClO2 et à l'acide acétique (pH entre 4,2 et 4,7) pendant 24 h à 75 °C (Sun et al., 1995). D'autres agents de délignification existent comme l'acide peracétique qui est sélectif de la lignine (Teixeira et al., 1999), le H2O2 en milieu alcalin (Fang et al., 1999) et le KMnO4 utilisé par Van Soest et al. (1985). Le KMnO4 est un agent d'oxydation plus fort que le NaClO2 (Collings et al., 1978). Afin d'obtenir un résidu insoluble de cellulose purifiée contenant encore des cendres, les hémicelluloses résiduelles sont extraites à la KOH 24 % et à l'acide borique 2 % pendant 2 h et à 20 °C (Sun et al., 1995).
37Xylanes et xyloglucanes. Les méthodes de Cutillas-Iturralde et al. (1998) et de Genestie (2006) purifient à partir, respectivement, des parois de kaki et des sons de céréale, les xylanes et les xyloglucanes par voie chimique et chromatographique.
38L'échantillon est soumis à la délignification au NaClO2 40 % et au H2SO4 0,3 % pendant 2 h à 70 °C. Les xylanes et les xyloglucanes du culot obtenus après centrifugation sont extraits à la KOH 1 M, au NaBH4 20 mM et sous atmosphère d'azote pendant 2 h à 60 °C. Cette suspension est centrifugée afin de récupérer les xylanes dans le surnageant et les xyloglucanes dans le culot. Les xyloglucanes du culot sont extraits par quatre traitements à la KOH 4 M et au NaBH4 20 mM sous atmosphère d'azote pendant 24 h à 20 °C. La suspension résultante est centrifugée. Les quatre surnageants sont combinés pour obtenir la fraction des xyloglucanes. Les fractions de xylanes et de xyloglucanes sont purifiées par l'hydrolyse de l'amidon et des protéines par respectivement des glycohydrolases (α-amylase et pullulanase) et des protéases. Les enzymes sont désactivées et la solution est dialysée et centrifugée pour récupérer le surnageant. Les xylanes ou les xyloglucanes sont séparés des pectines par une chromatographie préparative d'échange d'anions sur une colonne de DEAE-Trisacryl (Cutillas-Iturralde et al., 1998 ; Genestie, 2006).
39β-glucanes et xylanes. La méthode d'Izydorczyk et al. (1998a et 1998b) extrait par voie enzymatique à partir de grains d'orge une fraction de β-glucanes et de xylanes. Les β-glucanes sont ensuite séparés des xylanes par une précipitation sélective au (NH4)2SO4.
40L'échantillon est traité dans de l'eau à 40 °C pendant 30 min. Il est soumis à deux centrifugations.
41Premièrement, les protéines du mélange des deux surnageants contenant les β-glucanes et les xylanes hydrosolubles sont dénaturées en chauffant la solution à 95 °C. L'élimination des protéines du mélange des deux surnageants est effectuée par filtration sur de la diatomite suivie par adsorption des protéines du filtrat sur de l'argile. Un nouveau surnageant est obtenu par centrifugation de la suspension. Ce surnageant est soumis à l'hydrolyse de l'amidon par de l'α-amylase pancréatique. Afin d'obtenir les β-glucanes et les xylanes hydrosolubles à purifier, la solution est dialysée pour en éliminer les hydrates de carbone non structuraux, chauffée à 95 °C pour en désactiver les enzymes ajoutées et centrifugées.
42Deuxièmement, les β-glucanes et les xylanes non hydrosolubles contenus dans le culot initial sont extraits par voie alcaline par une solution saturée en Ba(OH)2 contenant 1 % (p/v) de NaBH4. Les β-glucanes et les xylanes sont séparés du résidu insoluble par deux centrifugations. Les deux surnageants sont combinés et neutralisés par de l'acide acétique. Cette solution contenant les β-glucanes et les xylanes non hydrosolubles est soumise à l'hydrolyse de l'amidon et des protéines par respectivement de l'α-amylase pancréatique et de la pronase. Afin d'obtenir les β-glucanes et les xylanes non hydrosolubles purifiés, cette solution est dialysée pour en éliminer les hydrates de carbone non structuraux et les protéines hydrolysées, chauffée à 95 °C pour en désactiver les enzymes et centrifugée.
43Les extraits de β-glucanes et de xylanes hydrosolubles et non hydrosolubles isolés et à purifier subissent ensuite le même processus. Les extraits secs sont dissous dans un tampon phosphate à pH 7 et du (NH4)2SO4 est lentement ajouté jusqu'à 28 % de saturation. Après avoir laissé cette solution agir pendant une nuit à 5 °C, elle est centrifugée afin d'isoler le précipité. Le (NH4)2SO4 résiduel du surnageant est dialysé. Une fois le surnageant lyophilisé, le même procédé est répété mais en atteignant à chaque fois un pourcentage supérieur au précédent en (NH4)2SO4 et cela, jusqu'à saturation. Si le pourcentage de (NH4)2SO4 par rapport à la saturation est inférieur à 45 %, alors seuls les β-glucanes sont précipités. Au-delà de 45 %, les xylanes précipitent également et cela augmente avec le pourcentage de (NH4)2SO4 par rapport à la saturation (Izydorczyk et al., 1998a ; Izydorczyk et al., 1998b).
44Mannanes. Pour des extraits d'hémicelluloses hydrosolubles ou des hémicelluloses extraites par voie alcaline venant de bois de gymnospermes et purifiés des pectines par une chromatographie préparative d'échange d'anions, les mannanes peuvent être précipités sélectivement par rapport aux autres polysaccharides structuraux par voie chimique.
45Les mannanes sont extraits dans l'eau ou à la NaOH diluée. La précipitation des mannanes est ensuite réalisée par du Ba(OH)2 0,03 M. Le précipité est récupéré par centrifugation et est purifié par la même étape de précipitation que celle décrite ci-dessus. Le nouveau précipité de mannanes obtenus par centrifugation est extrait à l'acide acétique 2 M et reprécipité à l'éthanol (Selvendran et al., 1985).
46Pectines. La méthode de Le Goff et al. (2001) fractionne les pectines de gousse de pois en trois catégories : solubles en milieu alcalin, solubles en milieu acide et solubles en milieu neutre.
47Les pectines sont déestérifiées en milieu alcalin par un traitement à la NaOH 1 M pendant 2 h à 4 °C. Ce traitement permet également d'extraire la fraction des substances, dont les pectines, solubles en milieu alcalin. Afin de faire descendre le pH à 5, de l'acide acétique glacial est ajouté à la solution. La suspension résultante est ensuite filtrée afin d'obtenir la fraction des substances, dont les pectines, extraites en milieu alcalin dans le filtrat. La fraction des substances, dont les pectines, solubles en milieu acide est extraite du résidu insoluble au HCl 1 M pendant 24 h à 80 °C. La suspension résultante est ensuite centrifugée afin d’obtenir dans le surnageant la fraction des substances, dont les pectines, solubles en milieu acide. La fraction des substances, dont les pectines, solubles en milieu neutre, est extraite du résidu insoluble en ramenant le pH à 7. Cette fraction est centrifugée afin d'isoler les substances, dont les pectines, solubles en milieu neutre dans le surnageant. Les trois fractions de pectines extraites sont purifiées des autres substances extraites comme les polysaccharides neutres par une chromatographie préparative d'échange d'anions sur une colonne de DEAE-Sepharose CL-6B (Le Goff et al., 2001).
48Les polysaccharides acides (les pectines acides et les glucuronoxylanes) peuvent être précipités par rapport aux polysaccharides neutres par une concentration en Ba(OH)2 de 0,15 M (Selvendran et al., 1985).
3. Comparaison des méthodes globales d'analyse des polysaccharides structuraux des biomasses lignocellulosiques
49Les méthodes d'analyse des polysaccharides structuraux des biomasses lignocellulosiques décrites ci-dessus sont basées sur un ou des objectifs précis à atteindre, qui sont propres à chaque méthode. Chaque méthode est fondée sur une ou plusieurs hypothèses et néglige certains facteurs qui n'ont pas d'intérêt dans le domaine d'application pour lequel elle a été initialement optimisée.
50Les méthodes globales comme celles de Van Soest, de Prosky, des polysaccharides non amylacés et d'Uppsala permettent la quantification de fractions insolubles. Ces fractions insolubles peuvent contenir un ou plusieurs types de polysaccharides structuraux des biomasses lignocellulosiques dont la nature est connue. Ces méthodes globales ne renseignent pas sur la quantité relative d'un type de polysaccharide structural par rapport aux autres types de polysaccharides structuraux contenus dans la même fraction des biomasses lignocellulosiques. Les méthodes quantifiant les polysaccharides structuraux des biomasses lignocellulosiques par gravimétrie (méthodes de Van Soest et de Prosky) (Tableau 1) ne donnent aucune information sur la composition en monosaccharides des polysaccharides structuraux. Ces deux méthodes se limitent simplement à déterminer la quantité de fibres alimentaires en y intégrant la lignine et les tannins insolubles à pH neutre. Les fractions isolées contiennent également des protéines insolubles au détergent neutre, des protéines insolubles au détergent acide ou non enzymatiquement hydrolysables, des pertes de diatomite (dans la méthode de Prosky) et des cendres (Prosky et al. 1985 ; Van Soest et al., 1985 ; Van Soest et al., 1991 ; Prosky et al., 1992), que l'analyse prend en compte par des dosages séparés. Pour les méthodes de quantification chromatographique et spectrophotométrique (méthode des polysaccharides non amylacés, d'Uppsala et de Selvendran) (Tableau 1), les valeurs obtenues pour les polysaccharides structuraux des biomasses lignocellulosiques sont corrigées par les facteurs de dégradation des monosaccharides libérés pendant l'hydrolyse acide des polysaccharides en monosaccharides et les facteurs de conversion des polysaccharides en monosaccharides (Englyst et al., 1994 ; Theander et al., 1995).
51En plus de donner la nature des monosaccharides constituant les polysaccharides structuraux par chromatographie et spectrophotométrie, la méthode de Selvendran fractionne sélectivement les polysaccharides structuraux des biomasses lignocellulosiques en fonction de leur nature, c'est-à-dire les pectines de la lamelle mitoyenne, les pectines de la paroi primaire, les xylanes, les xyloglucanes, les mannanes et la cellulose (Selvendran et al., 1985). Les méthodes de Van Soest et des polysaccharides non amylacés permettent également de quantifier la cellulose (Van Soest et al., 1991 ; Englyst et al., 1994).
52Selon la méthode globale choisie, les polysaccharides structuraux isolés sont différents pour une biomasse lignocellulosique donnée (Tableau 2).
53La méthode de Van Soest ne prend pas en compte les pectines, les gommes et les mucilages dans les fibres alimentaires, sauf si les ADF n'ont pas subi de traitement au détergent neutre. Dans ce cas, certaines pectines sont comprises dans le dosage des ADF (Van Soest et al., 1991 ; Ampuero, 2008). Dans le cas des méthodes de Prosky, des polysaccharides non amylacés et d'Uppsala, les pectines, les gommes et les mucilages font partie des fibres alimentaires. Les fibres alimentaires peuvent être séparées en fibres solubles et fibres insolubles dans l'eau, mais sans que les polysaccharides constituant chaque fraction soient isolés les uns des autres. Les pectines, les mucilages et les gommes très hydrosolubles ne sont pas précipités par l'éthanol et ne font donc pas partie des fibres alimentaires (Asp, 1995). Dans la méthode de Selvendran, les pectines sont extraites sélectivement en pectines de la lamelle mitoyenne et en pectines de la paroi primaire. Les mucilages et les gommes hydrosolubles ont une solubilité dans l'eau similaire aux pectines. Si ces mucilages et ces gommes sont présents dans un échantillon, alors ils se retrouveront fort probablement dans les fractions pectiques. Les mucilages et les gommes neutres peuvent être séparés des pectines par chromatographie d'échange d'anions. S'ils sont acides, leur séparation par rapport aux pectines dépend de leur teneur en acides uroniques.
54Dans la méthode des polysaccharides non amylacés, la précipitation des fibres alimentaires à l'éthanol acidifié à pH 2 conserve dans la fraction extraite les mono/di/oligo-saccharides et l'acide galacturonique libre. Cela n'est pas le cas pour la précipitation à l'éthanol de Prosky et d'Uppsala où le pH est supérieur à 2 (Englyst et al., 1994).
55L'amidon, polysaccharide non structural, est considéré de différentes manières selon les méthodes. Les méthodes de Van Soest, des polysaccharides non amylacés et de Selvendran éliminent tout l'amidon, alors que celles de Prosky et d'Uppsala hydrolysent l'amidon non résistant, RS1 et RS2 mais pas le RS3 qui est donc compris dans les fibres alimentaires (Van Soest et al., 1985 ; Van Soest et al., 1991 ; Asp, 1995 ; Englyst et al., 1996 ; FAO, 2007).
56Les méthodes globales peuvent être résumées de la manière suivante :
57– La méthode de Van Soest (Figure 2) ne prend pas en compte les pectines, les gommes et les mucilages, ne donne pas d'information sur la nature d'hémicelluloses contenues dans les NDF et fournit des données comme la quantité de cellulose, d'hémicelluloses et de lignine avec des tannins insolubles à pH acide. Les fractions de NDF et d'ADF ne contiennent pas de polysaccharides non structuraux. Cette méthode est utilisée dans le cadre de la quantification des fibres alimentaires pour les ruminants, ainsi que pour les non-ruminants si l'aliment ne contient pas trop d'amidon. Dans le cas contraire, la variante de Mertens (2002) de la méthode de Van Soest est employée (voir le point 2.1. pour plus de détails).
58– La méthode de Prosky (Figure 3) extrait des fractions qui contiennent des polysaccharides structuraux, des polysaccharides non structuraux (mucilages, gommes et amidon RS3) et d'autres composés comme de la lignine, des tannins, des protéines et des cendres. Elle ne donne aucune information sur la structure (cellulose, hémicelluloses et pectines) et la composition en monosaccharides des polysaccharides structuraux isolés des fibres alimentaires. La seule information disponible par cette méthode est le caractère soluble ou insoluble des fibres alimentaires dans l'eau à pH neutre. Cette méthode est utilisée dans le domaine du dosage des fibres alimentaires en alimentation humaine.
59– La méthode des polysaccharides non amylacés (Figure 4) extrait des fractions qui contiennent des polysaccharides structuraux, dose la cellulose, quantifie les monosaccharides constituant les polysaccharides structuraux isolés et sépare les polysaccharides non amylacés solubles et insolubles dans l'eau à pH neutre. Elle ne permet pas de différencier les hémicelluloses, les pectines, les gommes et les mucilages dans les fractions extraites. Cette méthode est employée afin de doser les polysaccharides non amylacés sans ambigüité.
60– La méthode d'Uppsala (Figure 5) isole des fractions formées des mêmes constituants que celle de Prosky. La méthode d'Uppsala quantifie les monosaccharides constituant les polysaccharides isolés. Dans la fraction insoluble, cette méthode dose la lignine de Klason. Elle ne permet pas de différencier la cellulose, les hémicelluloses, les pectines, les gommes et les mucilages dans les fractions extraites. Cette méthode est utilisée dans le contexte des fibres alimentaires en alimentation humaine.
61– La méthode de Selvendran (Figure 6) extrait sélectivement les pectines avec les mucilages et les gommes, les xylanes, les xyloglucanes, les mannanes et la cellulose. Les polysaccharides neutres peuvent être séparés des polysaccharides acides par une chromatographie d'échange d'anions. Elle permet de quantifier les monosaccharides constituant les polysaccharides structuraux isolés. Cette méthode extrait sélectivement les principaux types de polysaccharides structuraux et en caractérise la composition. Du fait du nombre important d'étapes d'extraction, l'analyse d'un échantillon est de longue durée, mais apporte une quantité d'information importante concernant les polysaccharides structuraux.
62Le classement des méthodes en ordre croissant des données qu'elles apportent sur la nature des polysaccharides structuraux des biomasses lignocellulosiques est le suivant : la méthode de Prosky, d'Uppsala, des polysaccharides non amylacés, de Van Soest et de Selvendran. Le classement des méthodes en ordre croissant du temps d'analyse pour l'analyse des polysaccharides structuraux des biomasses lignocellulosiques est le suivant : la méthode de Prosky, des polysaccharides non amylacés, d'Uppsala, de Van Soest et de Selvendran. La méthode de Van Soest présente à notre avis le meilleur compromis entre l'analyse des polysaccharides structuraux constituant la biomasse lignocellulosique et le temps d'analyse pour parvenir à obtenir cette information. Elle permet d'isoler en un minimum de temps des fractions dont trois qui sont majoritairement composées respectivement des pectines-mucilages-gommes, d'hémicelluloses et de cellulose. La fraction contenant les pectines-mucilages-gommes n'est pas quantifiée par cette méthode. Les pectines de cette fraction pourraient être séparées des mucilages et des gommes neutres par chromatographie d'échange d'anions. Si les gommes et les mucilages sont acides, leur séparation par rapport aux pectines dépend de leur teneur en acides uroniques. L'acide galacturonique, un acide uronique constituant majoritaire des pectines, pourrait être quantifié dans la fraction de pectines purifiées en appliquant le protocole de quantification des acides uroniques par HPLC de la méthode des polysaccharides non amylacés. Pour apporter des données sur la composition en monosaccharides des polysaccharides structuraux des biomasses lignocellulosiques, la méthode de Van Soest pourrait être mise en œuvre pour préparer les différentes fractions qui pourraient ensuite être hydrolysées en vue d'une quantification des monosaccharides par chromatographie analytique (comme dans la méthode des polysaccharides non amylacés et d'Uppsala). La méthode de Van Soest pourrait aussi apporter des données sur les hydrates de carbone non structuraux (mono/di/oligo-saccharides, amidon et fructanes) des biomasses lignocellulosiques, si ceux-ci sont isolés préalablement à l'extraction au détergent neutre. Afin de réaliser cette extraction des hydrates de carbone non structuraux des biomasses lignocellulosiques, le protocole de Hoebregs (1997) ou celui de McCleary (2004) pourraient être adaptés et appliqués. Ces méthodes permettent d'extraire les mono/di/oligo-saccharides, les fructanes et l'amidon des biomasses lignocellulosiques à l'aide d'eau à pH neutre à 85 °C pendant 10 min selon le protocole de Hoebregs ou à 80 °C pendant 15 min selon le protocole de McCleary.
4. Conclusion
63Vu la diversité et la complexité des polysaccharides structuraux contenus dans les biomasses lignocellulosiques, la détermination de leur structure et de leur composition en monosaccharides est à effectuer par une méthode éliminant les hydrates de carbone non structuraux ainsi que la lignine et caractérisant sélectivement les polysaccharides structuraux dont les fractions isolées sont hydrolysées, puis analysées par chromatographie analytique. Les fractions extraites peuvent éventuellement être purifiées par chromatographie préparative d'échange d'anions afin de séparer les polysaccharides neutres des polysaccharides acides. Pour les échantillons fortement lignifiés, afin de rendre le réseau de cellulose et d'hémicelluloses plus accessible à l'extraction des polysaccharides structuraux, il est préférable d'intégrer une étape de délignification dans la méthode de fractionnement.
64La méthode de Van Soest est la méthode présentant à notre avis le meilleur compromis entre les données obtenues sur la nature des polysaccharides structuraux constituant les biomasses lignocellulosiques et le temps d'analyse pour parvenir à obtenir cette information. En adaptant cette méthode, elle pourrait contribuer à apporter des données sur la quantité de pectines (exprimée en acide galacturonique), sur la composition en monosaccharides des polysaccharides structuraux et sur les hydrates de carbone non structuraux.
65Actuellement, il n'existe pas assez de données pertinentes dans la littérature comparant l'utilisation des méthodes globales présentées sur un même échantillon ou sur des échantillons semblables de biomasse lignocellulosique.
66Liste des abréviations
67ADF : fibres insolubles au détergent acide
68AOAC : Association of Official Analytical Chemists
69CDTA : N,N,N',N'-tétraacétate cyclohexanediamine
70DMSO : diméthylsulfoxide
71GC : chromatographie gazeuse
72HPLC : chromatographie liquide à haute performance
73NDF : fibres insolubles au détergent neutre
74SDC : déoxycholate de sodium
75SDS : dodécyl sulphate de sodium
76RS1 : amidon résistant de type 1
77RS2 : amidon résistant de type 2
78RS3 : amidon résistant de type 3
Bibliographie
Ampuero S., 2008. Détermination de la teneur en fibres dans les aliments pour animaux à ALP. Posieux, France : Agroscope Liebefeld-Posieux (ALP).
AOAC, 1990. Official methods of analysis. 15th ed. Washington, DC, USA: Association of Official Analytical Chemist.
Asp N.-G., 1995. Dietary fibre analysis. An overview. Eur. J. Clin. Nutr., 49(Suppl. 3), S42-S47.
Carpita N. & McCann M., 2000. The cell wall. In: Buchanan B., Gruissem W. & Jones R., eds. Biochemistry and molecular biology of plants. Rockville, USA: American Society of Plant Physiologists, 52-108.
Chai W. & Udén P., 1998. An alternative oven method combined with different strengths in the analysis of neutral detergent fibre. Anim. Feed Sci. Technol., 74, 281-288.
Collings G., Yokoyama M. & Bergen W., 1978. Lignin as determined by oxidation with sodium chlorite and a comparison with permanganate lignin. J. Dairy Sci., 61, 1156-1160.
Cutillas-Iturralde A., Peña M., Zarra I. & Lorences E., 1998. Xyloglucan from persimmon fruit cell walls. Phytochemistry, 48(4), 607-610.
Dorleans M., Mandran N. & Sauvant D., 1996. Study of the use of a protease with the Van Soest procedure. Anim. Feed Sci. Technol., 61, 129-136.
Ebringerová A., Hromádková Z. & Heinze T., 2005. Hemicellulose. Adv. Polym. Sci., 186, 1-67.
Englyst H., Quigley M. & Hudson G., 1994. Determination of dietary fibre as non-starch polysaccharides with gas-liquid chromatographic, high-performance liquid chromatographic or spectrophotometric measurement of constituent sugars. Analyst, 119, 1497-1509.
Englyst H. & Hudson G., 1996. The classification and measurement of dietary carbohydrates. Food Chem., 57(1), 15-21.
Eriksson I., Andersson R. & Aman P., 1997. Extraction of pectic substances form dehulled rapeseed. Carbohydr. Res., 301, 177-185.
Fang J. et al., 1999. Comparative study of hemicelluloses from wheat straw by alkali and hydrogen peroxide extractions. Polym. Degrad. Stab., 66, 423-432.
FAO, 2007. Directives concernant l'utilisation des allégations relatives à la nutrition : projet de tableau des conditions applicables à la teneur en éléments nutritifs (Partie B : Fibres alimentaires) à l'étape 6. Rome : FAO.
Genestie B., 2006. Optimisation de la production d'arabinoxylooligosaccharides d'intérêt biologique à partir de sons de céréales : approches méthodologiques. Thèse de doctorat : Université de Limoges (France).
Giger-Reverdin S., 1995. Review of the main methods of cell wall estimation: interest and limits for ruminants. Anim. Feed Sci. Technol., 55, 295-334.
Grabber J., 2005. How do lignin composition, structure, and cross-linking affect degradibility? Crop Sci., 45, 820-831.
Hatfield R. et al., 1994. A comparison of the insoluble residues produced by the Klason lignin and acid detergent lignin procedures. J. Sci. Food Agric., 65, 51-58.
Hoebregs H., 1997. Fructans in foods and food products, ion-exchange chromatographic method: collaborative study. AOAC Int. J., 80(5), 1029-1037.
Izydorczyk M., Macri L. & MacGregor A., 1998a. Structure and physiochemical properties of barley non-starch polysaccharides - I. Water-extractable β-glucans and arabinoxylans. Carbohydr. Polym., 35, 249-258.
Izydorczyk M., Macri L. & MacGregor A., 1998b. Structure and physiochemical properties of barley non-starch polysaccharides - II. Alkali-extractable β-glucans and arabinoxylans. Carbohydr. Polym., 35, 259-269.
Jung H.-J., 1997. Analysis of forage fiber and cell walls in ruminant nutrition. J. Nutr., 127(5), 810S-813S.
Le Goff A., Renard C., Bonnin E. & Thibault J.-F., 2001. Extraction, purification and chemical characterisation xylogalacturonans from pea hulls. Carbohydr. Polym., 45, 325-334.
Lee S., Prosky L. & De Vries J., 1992. Determination of total, soluble, and insolubles dietary fiber in foods-enzymatic-gravimetric method, MES-TRIS Buffer: collaborative study. AOAC Int. J., 75(3), 395-416.
McCleary B. & Monaghan D., 2002. Measurement of resistant starch. AOAC Int. J., 85(2), 665-675.
McCleary B. & Rossiter P., 2004. Measurement of novel dietary fibers. AOAC Int. J., 87(4), 707-717.
Mertens D., 2002. Gravimetric determination of amylase-treated neutral detergent fiber in feeds with refluxing in beakers or crucibles: collaborative study. AOAC Int. J., 85(6), 1217-1240.
Morrison I., 1980. Hemicellulosic contamination of acid detergent residues and their replacement by cellulose residues in cell wall analysis. J. Sci. Food Agric., 31, 639-645.
Prosky L. et al., 1985. Determination of total dietary fiber in foods and food products: collaborative study. AOAC J., 68(4), 677-679.
Prosky L. et al.., 1992. Determination of insoluble and soluble dietary fiber in foods and food products: collaborative study. AOAC Int. J., 75(2), 360-367.
Selvendran R. & O'Neill M., 1985. Isolation and analysis of cell walls from plant material. In: Glick D., ed. Methods of biochemical analysis. Vol. 32. New York, USA: Wiley, 25-153.
Southgate D., 1995. Dietary fibre analysis. Cambridge, UK: The Royal Society of Chemistry.
Sun R., Lawther M. & Banks W., 1995. Influence of alkaline pre-treatments on the cell wall components of wheat straw. Ind. Crops Prod., 4, 127-145.
Teixeira L., Linden J. & Schroeder A., 1999. Alkaline and peracetic acid pretreatments of biomass for ethanol production. Appl. Biochem. Biotechnol., 77-79, 19-34.
Theander O. et al., 1995. Total dietary fiber determined as neutral sugar residues uronic acid residues, and klason lignin (the Uppsala method): collaborative study. AOAC Int. J., 78(4), 1030-1044.
Van Soest P. & Robertson J., 1985. Analysis of forages and fibrous foods. Ithaca, USA: Department of Animal Science, Cornell University.
Van Soest P., Robertson J. & Lewis B., 1991. Methods for dietary fiber, detergent fiber, and nonstarch polysaccharides in relation to animal nutrition. J. Dairy Sci., 74, 3583-3597.
Zhang Y.-H., 2008. Reviving the carbohydrate economy via multi-product lignocellulose biorefineries. J. Ind. Microbiol. Biotechnol., 35, 367-375.
Voetnoten
Om dit artikel te citeren:
Over : Bruno Godin
Centre wallon de Recherches agronomiques (CRA-W). Département Valorisation des Productions. Unité Biomasse, Bioproduits et Énergies. Chaussée de Namur, 146. B-5030 Gembloux (Belgique). E-mail : b.godin@cra.wallonie.be – Université catholique de Louvain (UCL). Unité de Génie biologique. Croix du Sud, 2/19. B-1348 Louvain-la-Neuve (Belgique).
Over : Richard Agneessens
Centre wallon de Recherches agronomiques (CRA-W). Département Valorisation des Productions. Unité Technologies de la Transformation des Produits. Rue du Serpont, 100. B-6800 Libramont (Belgique).
Over : Sébastien Gofflot
Centre wallon de Recherches agronomiques (CRA-W). Département Valorisation des Productions. Unité Technologies de la Transformation des Produits. Rue du Serpont, 100. B-6800 Libramont (Belgique).
Over : Stéphane Lamaudière
Université catholique de Louvain (UCL). Unité de Génie biologique. Croix du Sud, 2/19. B-1348 Louvain-la-Neuve (Belgique).
Over : Georges Sinnaeve
Centre wallon de Recherches agronomiques (CRA-W). Département Valorisation des Productions. Unité Technologies de la Transformation des Produits. Rue du Serpont, 100. B-6800 Libramont (Belgique).
Over : Patrick A. Gerin
Université catholique de Louvain (UCL). Unité de Génie biologique. Croix du Sud, 2/19. B-1348 Louvain-la-Neuve (Belgique).
Over : Jérôme Delcarte
Centre wallon de Recherches agronomiques (CRA-W). Département Valorisation des Productions. Unité Biomasse, Bioproduits et Énergies. Chaussée de Namur, 146. B-5030 Gembloux (Belgique).